


Introduction
Global developmental delay (GDD) is defined as a significant delay in two or more domains of development, including activities of daily living as well as motor, cognitive, speech/language, and personal/social skills1,2). The etiology of GDD is heterogeneous, and its diagnosis has been proven to be very difficult; thus, the causes of GDD are undetermined in approximately 62% of children3). Individuals in whom genetic causes are established generally undergo very exhaustive, expensive, and often invasive diagnostic evaluations, even though the results may not change the medical or therapeutic management of the delay. Therefore, a diagnostic approach focusing on treatment has been proposed as an alternative for patients diagnosed with GDD. Inborn errors of metabolism (IEM) are uncommon causes of GDD; however, metabolic evaluations of GDD are an important step because there is an increasing availability of treatments for metabolic conditions, which may lead to better clinical outcomes. A recent study with next-generation sequencing analysis (NGS) revealed that the clinical phenotypes of many genetic or metabolic causes of GDD overlap, making diagnosis difficult and challenging. Therefore, evidence-based cost-effective strategies for the identification of treatable conditions at the early stages of GDD evaluation may be helpful in clinical practice, particularly where financial support is limited.
Causes of GDD
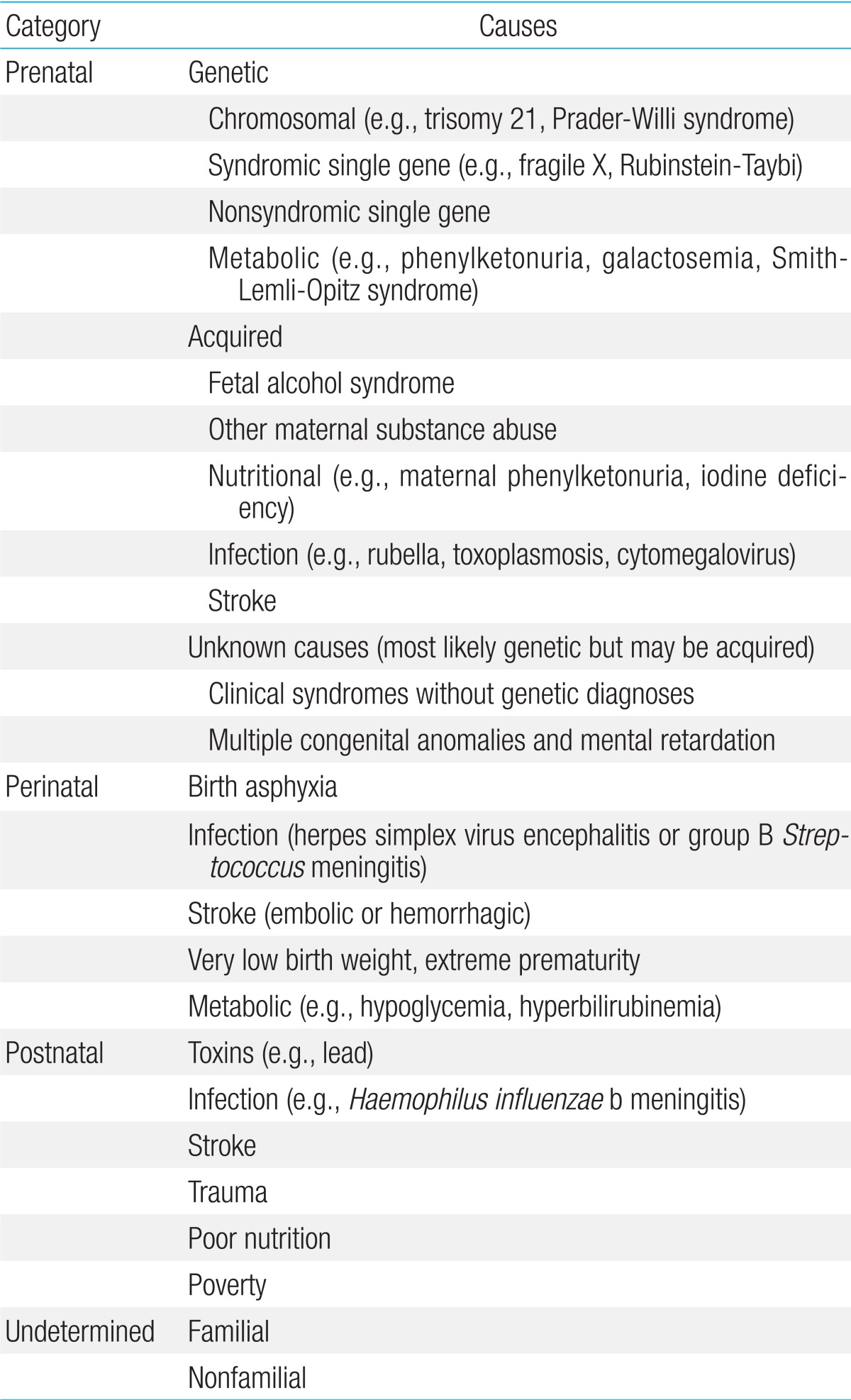
GDD may be caused by prenatal, perinatal, postnatal, or undetermined causes (Table 1)4). There are a large number of known specific causes of GDD, of which genetic or chromosomal causes constitute the largest category. Despite our considerable knowledge on the potential causes of GDD, the specific origin remains unknown in many individuals, although genetic causes have been increasingly recognized in patients with GDD. Clinical diagnostic tests using NGS have been implemented in several diagnostic reference laboratories in the United States.
Approaches for children with GDD
The initial clinical assessment of children with GDD involves the evaluation of their medical history, including prenatal, perinatal, and postnatal histories; reviewing their family history of 3 generations or more; physical and neurological examinations; and a thorough assessment of behavioral signs that may indicate a specific disease. The child's family history should be carefully obtained, while assessing whether other family members have similar or other potentially relevant neurological impairments. In addition, the mother's gestational history and pregnancy with the affected child (i.e., evidence of adverse events, toxin exposures, and intrauterine difficulties) and timing and mode of delivery (i.e., reasons for forceps application or Cesarean section) should be noted. Furthermore, the child's birth weight, Apgar scores, head circumference measurement at birth, and duration of postnatal hospital stay are important objective markers of newborn health status. Difficulties in the newborn nursery, particularly those due to possible neonatal encephalopathy (e.g., seizures, feeding difficulties prompting gavage), should be carefully documented. It is also important to obtain newborn screening (NBS) results. NBS panels that use tandem mass spectrometry dramatically improve early diagnosis and outcomes with appropriate treatments, but current NBS tests target only a minority of treatable IEM. Parental recall of motor skill development in the first year of life and speech/language milestones is important. The potential loss or regression of previously acquired developmental skills should be particularly noted because this indicates the need for a more urgent etiologic evaluation and follow-up. Current skill levels in various developmental domains together with the degree of independence in activities of daily life, scholastic history, and coexisting medical problems should be noted.
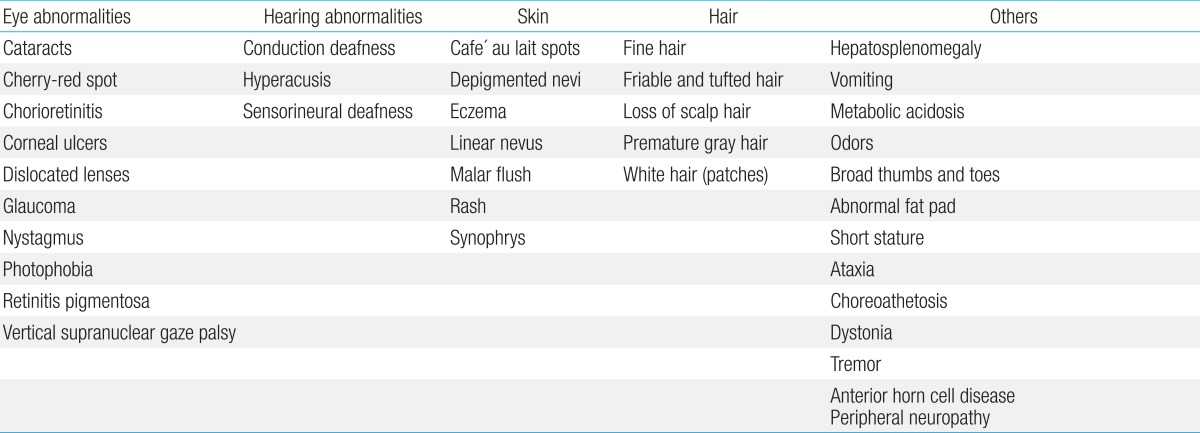
Physical examination should assess possible dysmorphology, hepatosplenomegaly, and cutaneous markers of neurocutaneous syndromes as well as the presence of dermal sinuses, hair tufts, or other subtle signs of spinal dysraphism in the sacral region. Furthermore, height, weight, and head circumference should be measured. Additional signs and symptoms can help to identify the causes of GDD (Table 2)4).
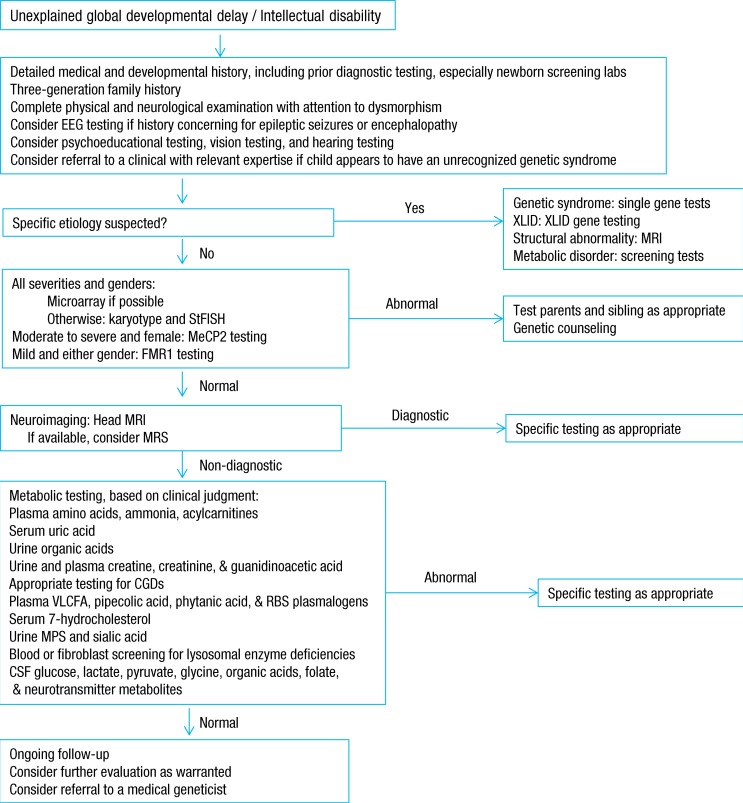
After clinical evaluation, appropriate diagnostic laboratory tests, brain imaging, and other evidence-based evaluations are applied for establishing diagnosis and for care planning5). With remarkable progress in the field of genetics over the last several years, chromosomal microarrays are now important first-line clinical diagnostic tests for children with GDD of unknown causes. Moreover, a renewed emphasis on the identification of "treatable" causes of GDD recommends screening for IEM in children with GDD of unknown causes4,5,6). The practice parameters and evidence report for GDD or intellectual disability published by the American Academy of Neurology provide a framework of recommendations for evaluation (Fig. 1). These include a combination of broad screening tests and disease-specific evaluations based on a heightened pretest probability (>1%), while considering the identified clinical features4).
Characteristics of developmental delay caused by IEM
Developmental delay is a major symptom of various IEM that appear at a young age. Therefore, understanding the general characteristics of developmental delay caused by IEM is an important step for better identification and appropriate follow-up with metabolic workup.
First, developmental delay caused by IEM is usually global, which implies that it affects, to some extent, all domains of development. Second, behavioral problems such as severe irritability, impulsivity, aggressiveness, and hyperactivity are common. Typical examples are Krabbe disease (globoid cell leukodystrophy), Sanfilippo disease (mucopolysaccharidosis type III [MPS III]), and Hunter disease (MPS II). Motor automatisms and stereotypic behaviors are also common in these disorders. Self-mutilation, such as compulsive chewing of the thumb and fingers, sometimes resulting in severe laceration of the lips and traumatic amputation of the fingers, is a prominent symptom in boys with Lesch-Nyhan syndrome. Third, delayed development is usually progressive in children with IEM. Developmentally delayed children with IEM may show a period of normal development, followed by a loss of developmental milestones. The progression may be subtle at first, but the gap between children with GDD and healthy children becomes wider with time. Finally, delayed development in children with IEM is usually associated with other objective neurological dysfunctions such as abnormal muscle tone, problems in specific senses, seizures, pyramidal tract signs, and extrapyramidal or cranial nerve deficits.
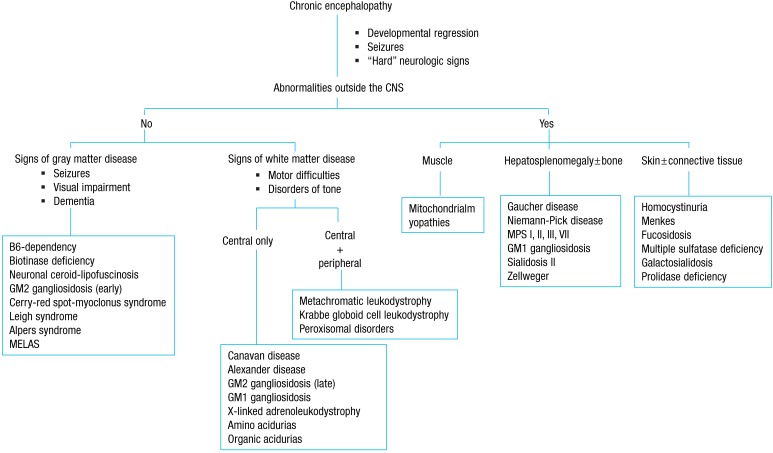
Age at onset and clinical course often provide important clues to the metabolic nature of the disease, and the involvement of other organs is frequent. The major affected areas outside the central nervous system include organs such as the liver and spleen, as well as the muscles, bones, skin, and connective tissues (Fig. 2). An initial evaluation for children with undifferentiated developmental delay includes thorough developmental assessment and neurological examination, brain imaging, electrophysiological studies (auditory brainstem responses, visual evoked potentials, somatosensory evoked potentials, nerve conduction studies, electromyogram), radiographs of the hands, chest, and lateral view of the spine for evidence of dysostosis multiplex, plasma amino acid analysis, urinary amino acid analysis, urinary organic acid analysis, even in the absence of overt metabolic acidosis, plasma ammonium, preferably two hours after a normal meal of protein-containing food, plasma lactate, urinary MPS screening, and urinary oligosaccharide screening test. It also includes tests for determining the extent and degree of neurological damage, ensuring that the early stages of some treatable IEM are not missed, and establishing a baseline for monitoring the natural history of the disease 7).
Diagnostic recommendations for the identification of treatable causes of GDD
If a diagnosis is unclear after a clinical assessment, a stepwise evaluation process is undertaken. At this stage, specific metabolic testing should be considered, including total serum homocysteine, acylcarnitine profile, amino acids, organic acids in urine, glycosaminoglycans, oligosaccharides, purines and pyrimidines, and guanidinoacetate-to-creatine ratio. Routine metabolic testing of children with GDD had a 0.8%-2.5% rate of diagnosis of IEM5,8,9), but a detailed metabolic reassessment identified the causative IEM in up to 14% of the patients previously classified as having GDD of an unknown cause10,11). These studies have raised the concern that treatable diagnoses may be missed if we focus too heavily on current practice parameters12,13).
In 2013, an updated systematic literature review identified 89 IEM, which present with GDD as a prominent feature and are amenable to causal therapy14). Most IEM are associated with additional neurological and/or nonneurological features. Neurological features include ataxia, behavioral disturbances, dementia, dystonia, encephalopathic crisis, epilepsy, hearing loss, hypotonia, myopathy, abnormalities in brain imaging (basal ganglia, cerebellum, cerebrum, white matter, and mixed), neuropathy, ocular movement abnormalities, psychiatric disturbances, sensorineural hearing loss, spasticity, stroke, or vision loss. Most IEM are associated with at least one additional prominent neurological feature, such as epilepsy and various types and degrees of movement disorders (e.g., spasticity, dyskinesia, and ataxia). However, many cases of IEM present with developmental delay as the sole feature for a considerable amount of time before manifestation of the full phenotype (e.g., disorders of creatine synthesis and transport). The nonneurological features affect the following anatomical/organ systems: bones and joints; skin and blood tissues; gastrointestinal, endocrine, and immune systems; organs including the eyes, liver, heart, and kidneys; and anatomical features including facial dysmorphism, growth, and stature.
These 89 IEM include disorders of specific molecules such as amino acids (n=12), cholesterol and bile acids (n=3), vitamins/cofactors (n=10); creatine (n=3), fatty aldehydes (n=1), purines and pyrimidines (n=3); metals (n=5), organic acids (n=19); disorders that affect cellular organelles such as lysosomes (n=12), mitochondria (n=2), and peroxisomes (n=1); anomalies in glucose homeostasis and transport (n=2), the urea cycle (n=8), neurotransmission (n=8), and hyperhomocysteinemia (n=7).
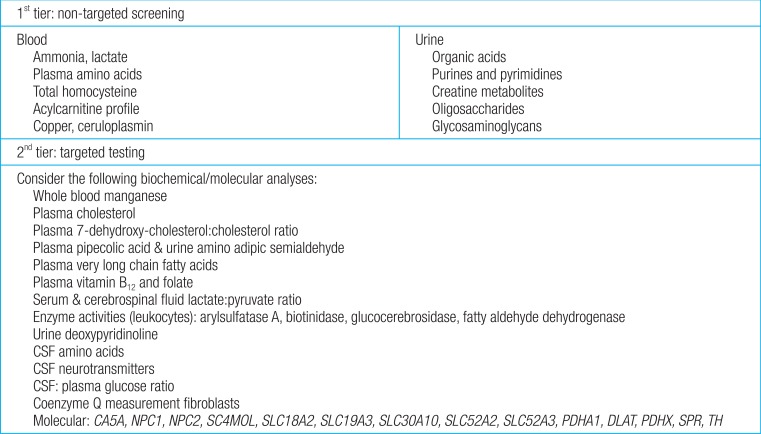
Fifty-four of the 89 disorders (60%) are identified by metabolic screening tests in blood (plasma amino acids, homocysteine, copper, and ceruloplasmin) and urine (creatine metabolites, glycosaminoglycans, oligosaccharides, organic acids, and pyrimidines). For the remaining 35 disorders (40%), specific tests are required, including primary molecular analysis. First-tier testing includes group metabolic tests in urine and blood, which should be performed in every patient with GDD of unknown cause. Based on the differential diagnosis generated by the patient's signs and symptoms, a second-tier test is prescribed individually using a low threshold. Selective metabolic testing should be performed if there are suggestive clinical findings, including lethargy, cyclic vomiting, early seizures, dysmorphic features, and developmental disabilities (Fig. 3)14).
Conclusions
Recent advances in genetic technology provide the possibility of improving diagnosis, and ultimately the outcomes, in patients with GDD. This could allow early diagnosis and the use of available treatments for the specific causes of GDD. Neurological pathologies are the most prominent clinical symptoms associated with many types of GDD, and, in particular, with IEM. However, general neurological issues are common, and deciding who should be evaluated and the type of testing needed is often challenging. Although metabolic disorders represent a small and extremely heterogeneous proportion of GDD cases, early diagnosis of metabolic disorders and proper therapeutic interventions may significantly improve the developmental outcomes in some of these patients. Therefore, the clinical application of innovative evidence-based evaluations for appropriate metabolic and genetic testing of GDD of unknown causes should be strongly considered.
 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


