




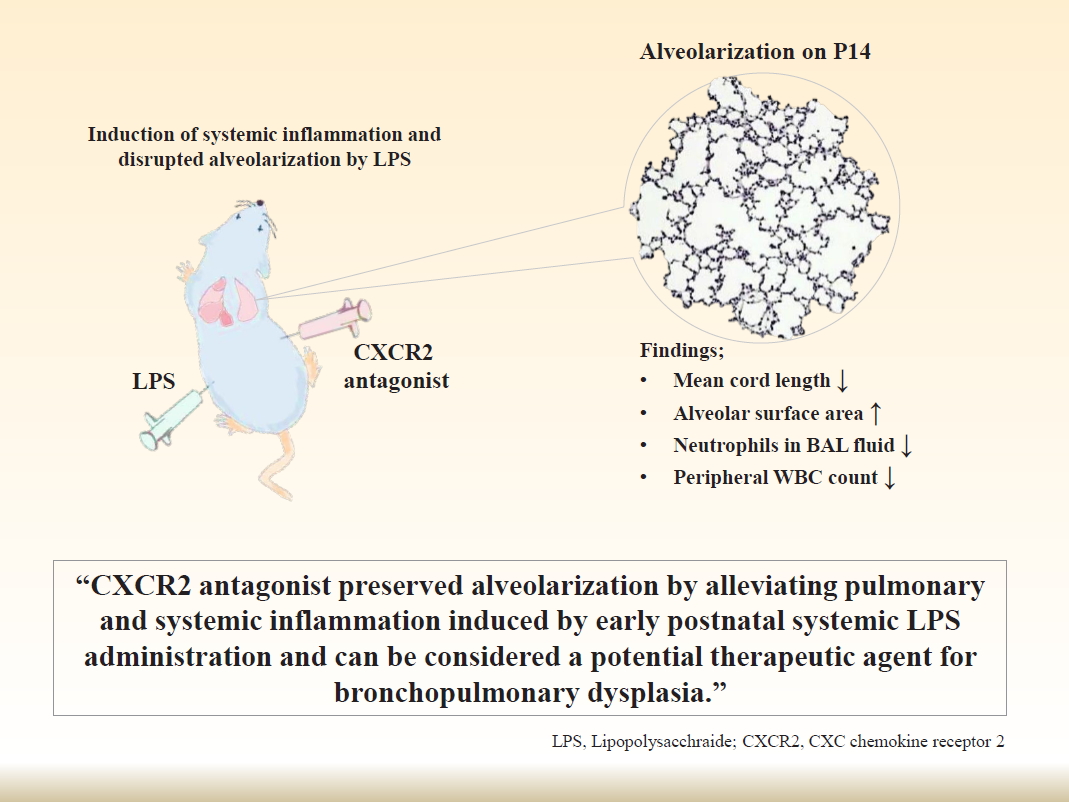
Graphical abstract.
Introduction
Bronchopulmonary dysplasia (BPD) is one of the major chronic complications suffered by surviving preterm infants and is associated with adverse pulmonary and neurodevelopmental outcomes [1]. Despite recent advances in perinatal and neonatal care, the incidence of BPD has changed little in the last 20 years [2,3]. In compliance with advances in neonatal medicine and clinical practices, the clinical manifestation and pathology of BPD have evolved from “old BPD” to “new BPD” [4]. “New BPD” is characterized by little damage to the airway epithelium, airway smooth muscle hyperplasia, and fibroproliferative changes, which are hallmarks of “old BPD”. Instead, alveolar and pulmonary vascular hypoplasia have become principal features of “new BPD” [5]. Inflammation plays a central role in the pathogenesis of “new BPD” [6]. Inflammation can be evoked by infectious organisms, oxidative stress, or mechanical ventilation [7].
Animal studies have shown that leukocytes influx precedes the development of BPD in premature sheep [8]. Neutrophils can exacerbate oxidant stress in an organ or local tissue through respiratory burst activation. Leukocyte depletion has been known to ameliorate free radical-mediated lung injury [9]. The influx of leukocytes into the site of inflammation is modulated by chemokines, which are released at the site of inflammation [10]. Chemokines are small, soluble peptides and interact with various cells through specific chemokine receptors. There are more than 40 members of chemokines and 19 different chemokines receptors [11]. CXC chemokines receptors (CXCR) 1 and CXCR2 are the major chemokines receptors of neutrophils [12]. Especially, CXCR2 has been implicated in numerous inflammatory diseases [13]. Selected blockage of CXCR2 in mice reduced neutrophil adhesive activity and lipopolysaccharide (LPS)-mediated neutrophil influx [14,15]. A competitive CXCR2 antagonist has led to a reduction in neutrophil sequestration and, thus neutrophil influx and neutrophil activation were avoided in local tissues [16]. The CXCR2 pathway has been implicated in the pathogenesis of BPD because of the predominance of CXCR2 ligands, such as interleukin (IL)-8 and growth-related oncogene-α in tracheal aspirates of preterm infants who developed BPD later [17,18].
In our previous study, we have demonstrated that early postnatal systemic inflammation induced by intraperitoneal (i.p.) LPS administration leads to an arrest in alveolarization in a newborn rat model [19]. In the present study, we further investigated whether a CXCR2 antagonist can preserve alveolarization by attenuating pulmonary and systemic inflammation induced by systemic LPS administration using the same newborn rat model.
Methods
1. Animal experiment
The animal experimental protocol was approved by the Seoul National University Bundang Hospital Institutional Animal Care and Use Committee (BA1201-096/003-01). On E20 (term 22.5 days), pregnant Sprague-Dawley rats were anesthetized by isoflurane inhalation. After making a midline abdominal incision, 0.1 mL of saline was injected into the amniotic sacs under direct visualization, because the control (CXCR2 antagonistuntreated) animals used in this experiment were shared with another experiment in which intra-amniotic LPS or saline was administered to the pregnant Sprague-Dawley rats on E20 [19]. After recovery, pups were spontaneously delivered 2–2.5 days after the injections. During the experiment, all pups were maintained under room air. At 10–12 hours after birth, rat pups were injected i.p. with either 0.25 mg/kg of LPS dissolved in 0.1 mL of saline or vehicle (saline) on P1, P3, and P5, which encompass the saccular and early alveolar stages of lung development in the rat [20]. In the same time with the administration of LPS or saline, 1 mg/kg of nonpeptide CXCR2 antagonist (SB-265610, Sigma-Aldrich, St. Louis, MO, USA) dissolved in 0.1 mL of dimethyl sulfoxide or vehicle (saline) was injected i.p. to rat pups on P1, P3, and P5. This experimental protocol led to 4 treatment groups: (1) vehicle and vehicle group (V+V); (2) LPS and vehicle group (LPS+V); (3) vehicle and CXCR2 antagonist group (V+C); (4) LPS and CXCR2 antagonist group (LPS+C). Throughout the experiment, each rat dam reared pups that were allocated to a single group only. A total of 141 fetal rats from 11 pregnant rats were used in the experiment.
2. Bronchoalveloar lavage fluid and peripheral blood collection and white blood cell and neutrophil counts
On P7 and P14, bronchoalveolar lavage fluid (BALF) and peripheral blood (PB) were collected from the rat pups. Five to six rat pups per group were used for BALF collection at each time point. To obtain differential counts, 200–300 cells were manually counted on each glass slide at ×200 magnification. The number of each white blood cell (WBC) type was calculated as the percentage of each WBC multiplied by the total number of WBCs in the BALF. For BALF collection, a different set of animals from those used for histological analysis was used to avoid possible interference of the BALF procedure with the histological analysis. PB was drawn directly from the left ventricle under anesthesia immediately before opening the chest wall for lung harvest on P7 and P14. Five to 6 rat pups per group were used to collect PB at each time point. The total WBC count of collected PB was measured with a Hemavet 950FS analyzer (Drew Scientific, Dallas, TX, USA). A differential cell count was manually performed on each PB smear slide at ×400 magnification using the same method as for BALF.
3. Evaluations for alveolarization
On P7 and P14, rat pups were sacrificed and their lungs were harvested and processed for the morphometric assessment. The detailed methods for lung harvest and processing are described in our previous study [19]. To assess alveolarization, 4 random, nonoverlapping fields per animal in 2 distal lung sections were captured at ×100 magnification. All measurements were made by a single observer unaware of group identities. The extent of alveolarization was objectively assessed using mean cord length (Lm) and alveolar surface area (SA). Lm is an estimate of the distance from one airspace wall to another airspace wall and was determined by counting the intersections of airspace walls, including the alveoli, alveolar sacs, and alveolar ducts, with an array of 84 lines, each ~24 μm long. Tissue volume density (VDT) was determined using a 10×10 grid (grid element side length ~29 μm). SA was calculated with the formula SA=4×VDTxlung volume/Lm. Six to seven rap pups were used to assess alveolarization at each time point.
Results
1. Survival rate
The survival patterns by the experimental group until P14 are presented in Fig. 1. Only 1 fetal death (1 of 141) occurred after intra-amniotic saline-administration. After birth, 54.2% (32 of 59) of the animals in the LPS+V group died until P7 with most death occurred after the first i.p. LPS injection. However, only 11.1% (3 of 27) of the animals in the V+V group died during the first 7 days after birth. The CXCR2 antagonist-treated groups had minimal mortality during the first 7 days after birth with only one death in the V+C group (1 of 23) and no death in the LPS+C group (0 of 31). There was no further mortality after P7 except for sacrificed animals for analyses in all 4 groups. Overall survival rate of the LPS+V group was significantly lower than all the other groups (P<0.001). Otherwise, there was no significant difference in overall survival rate among the groups. Twenty-four rat pups in the V+V group, 27 rat pups in the LPS+V group, 22 rat pups in the V+C group, and 31 rat pups in the LPS+C group survived until P7 and then were sacrificed for the analyses on P7 and P14.
2. WBC and neutrophil counts in BALF and PB
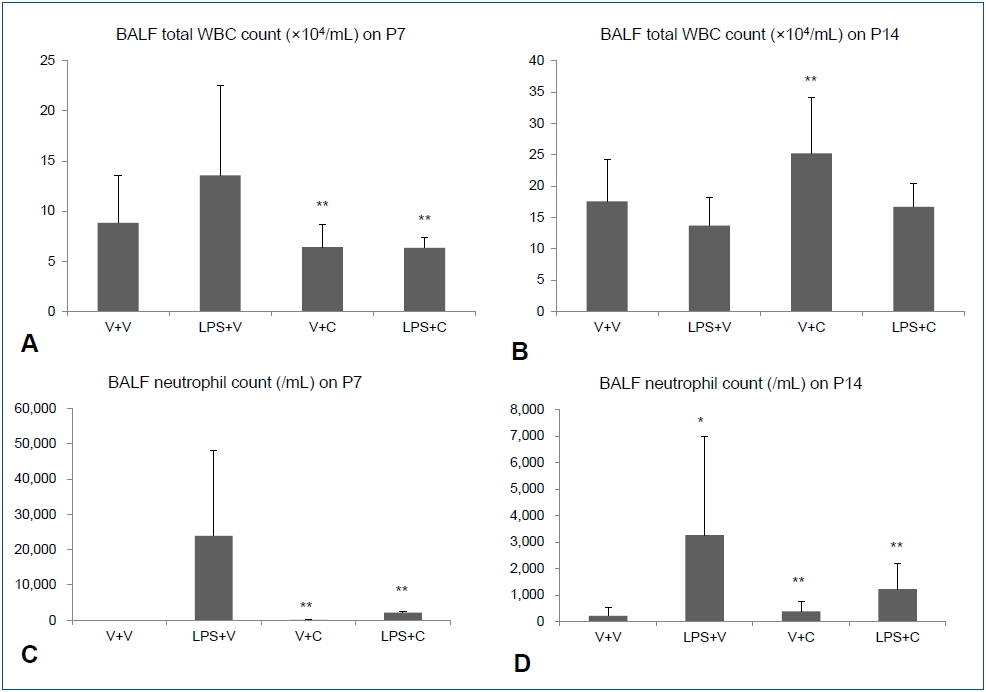
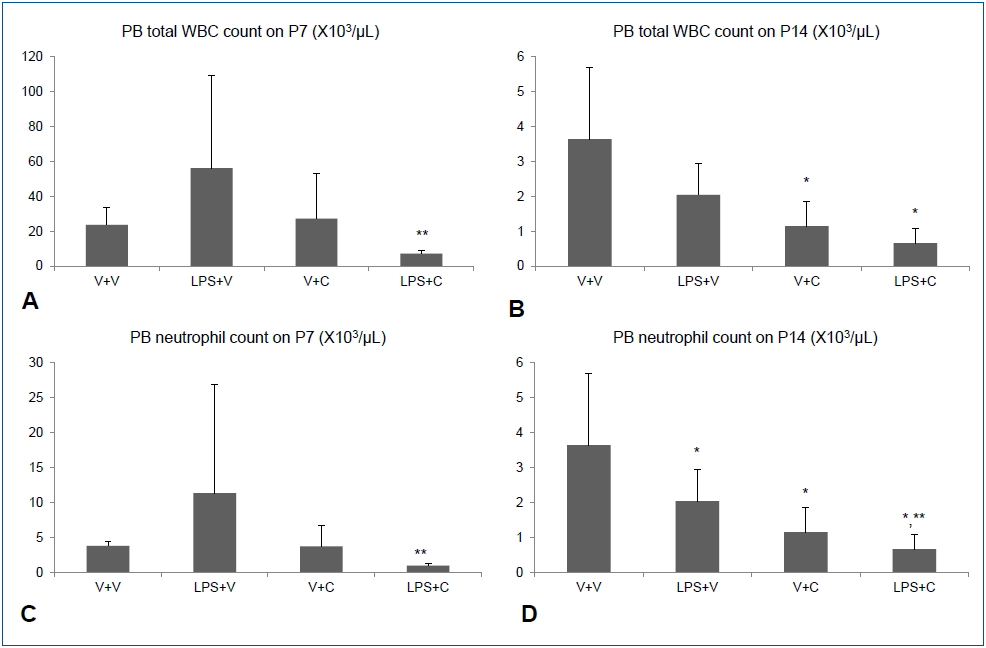
Intraperitoneal LPS increased total WBC and neutrophil counts in BALF on P7. Treatment with the CXCR2 with i.p. LPS led to significant lower total WBC and neutrophil counts in BALF than when i.p. LPS was treated with vehicle (Fig. 2A, C). On P14, there was no significant difference in total WBC count in BALF between the LPS+V group and the LPS+C group, but BALF neutrophil count was significantly higher in the LPS+V group than in the LPS+C group (Fig. 2B, D). Similarly, increased PB total WBC and neutrophil counts by i.p. LPS were significantly attenuated by CXCR2 antagonist treatment on P7 (Fig. 3A, C). On P14, PB total WBC and neutrophil counts were all markedly decreased compared to those on P7, although there were some statistically significant differences among the groups (Fig. 3B, D).
3. Alveolarization
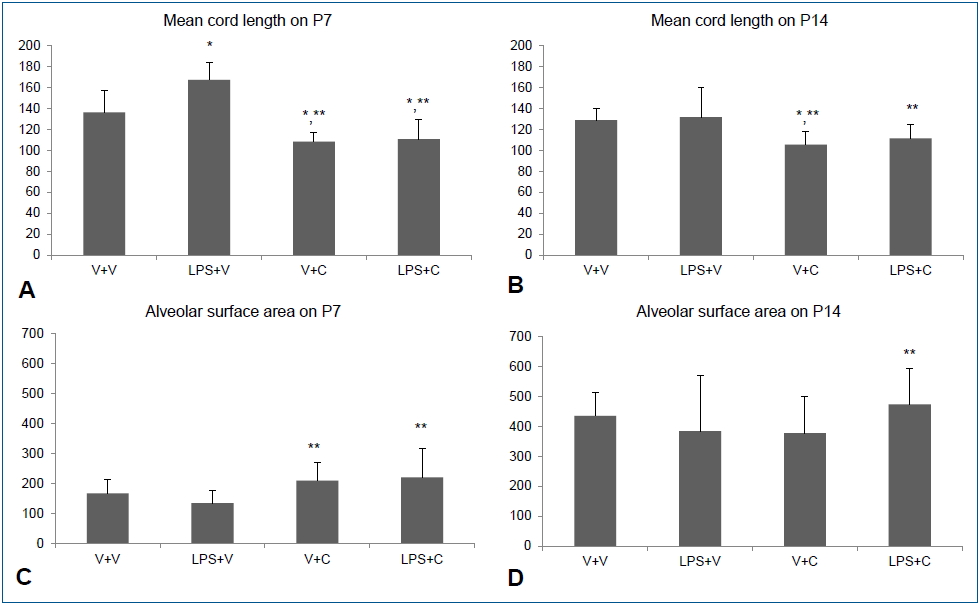
Intraperitoneal LPS led to large and simple distal air spaces which is a hallmark of disrupted alveolarization on P7. The alveolarization proceeded until P14, as indicated by small and complex distal air spaces in all groups except the LPS+V group. In the LPS+V group, the distal airspaces remained large and simple on P14, similar to their appearance on P7, indicating an arrest in alveolarization. Groups treated with CXCR2 antagonist showed normal alveolarization on both P7 and P14 (Fig. 4). The results of morphometric assessment were consistent with these microscopic findings. The Lm, which is an indicator of the average alveolar size, was significantly greater in the LPS+V group than in the V+V group and the CXCR2 antagonist-treated groups on P7 (Fig. 5A). On P14, Lm continued to be significantly greater in the LPS+V group than in the CXCR2 antagonist-treated groups (Fig. 5B). In contrast, the SA was significantly smaller in the LPS+V group than in the CXCR2-treated groups on P7 (Fig. 5C). On P14, while the SA increased in all groups as the lungs developed, the LPS+V group had significantly smaller SA than the LPS+C group (Fig. 5D).
Discussion
Recently, postnatal inflammation has been highlighted as one of major players in the development of BPD [21]. An influx of inflammatory cells and diverse neutrophil chemokines-derived inflammatory reactions have been implicated in BPD [8,17,22]. This study was designed to investigate whether CXCR2 antagonist can restore alveolarization by attenuating systemic and pulmonary inflammations induced by early postnatal systemic LPS administration. Because there was already a model of the interaction between hyperoxia and a CXCR2 antagonist, no pups were exposed to hyperoxia in the current study [23]. LPS was administered i.p. on P1, P3, and P5, which correspond to the saccular and early alveolar stages of lung development, to induce systemic inflammation [24]. In previous studies, postnatally administered LPS resulted in an overwhelming inflammatory response syndrome leading to high mortality [25,26]. Similarly, early postnatal i.p. LPS administration led to high mortality and systemic and pulmonary inflammation in the surviving rat pups in the present study. An arrest in alveolarization was demonstrated in these surviving rat pups. These findings suggest that early postnatal systemic inflammation is detrimental to alveolar development. As an intervention, the administration of CXCR2 antagonist together with LPS significantly attenuated the systemic and pulmonary inflammations. Furthermore, CXCR2 antagonist ameliorated the detrimental effect of early postnatal i.p. LPS on alveolarization and preserved normal alveolarization.
In a murine model of BPD induced by IL-1β expression, the knockout of CXCR2 led to opposite outcome depending on the timing of IL-1β expression [27]. When IL-1β was expressed from the pseudoglandular to alveolar stages, the deletion of CXCR2 led to reduced alveolarization and survival of the mice. However, the same deletion of CXCR2 resulted in improved alveolarization and survival of the mice, when IL-1β was expressed during the saccular stage. As described above, LPS and CXCR2 antagonist were administered simultaneously during the saccular and early alveolar stages of lung development in the present study. It remains to be revealed whether the protective effect of CXCR2 is also case before the saccular stage.
Macrophages in the injured tissue induce neutrophil influx through the expression of CXC [28,29]. Neutrophils are an essential component of the innate immune system and are the first group of cells that migrate into the site of infection or inflammation. CXCR1 and CXCR2 are the major chemokine receptors of neutrophils [30]. Just as competitive antagonism of the CXCR2 leads to a reduction in neutrophil sequestration and neutrophil influx, neutralization of CXCR2 ligands prevents neutrophil accumulation, as shown in several studies on acute and sub-acute inflammatory lung injuries [16,22]. Furthermore, selective inhibition of neutrophil function is a feasible strategy to reduce the adverse effects of inflammatory processes, and will reduce the need for glucocorticoid therapy. Because, postnatal glucocorticoid therapy is associated with an adverse long-term neurodevelopmental outcome in preterm infants, other selective anti-inflammatory strategies are required [31]. In this regard, blockage of CXCR2 would be a promising anti-inflammatory strategy for premature newborns who are at a greater risk of developing BPD.
Of noteworthy is that CXCR2 antagonist remarkably reduced mortality of rat pups exposed to postnatal systemic LPS. While the LPS+V group showed a substantial mortality during the first 3 days, the LPS+C group had no mortality until P14. This finding might be attributable to a strong anti-inflammatory action of CXCR2 antagonist against overwhelming systemic inflammatory responses induced by systemic LPS administration. However, other mechanisms than anti-inflammatory action can be involved in that substantially reduced mortality.
Most of murine models of BPD involve an exposure of hyperoxia [32]. In our previous study, we established a newborn rat model of BPD by combining intra-amniotic LPS administration and postnatal hyperoxic exposure [33]. In the present study, we used newborn rat model which involves only early postnatal LPS administration without an exposure to hyperoxia or intraamniotic LPS. Previously, we proved that early postnatal LPS administration alone can induce an arrest in alveolarization, a hallmark of BPD [19]. Because, the aim of the present study was to investigate the potential anti-inflammatory effect of CXCR2 antagonist in the early postnatal period, we chose to use a BPD model which involves only early postnatal LPS administration. This newborn rat model of BPD could be used to explore the mechanisms regarding how various inflammatory stimuli alter normal lung development and to discover new therapeutic approaches to BPD.
In the present study, we measured only total WBC and neutrophil counts as the inflammatory markers. Other inflammatory markers such as C-reactive protein, proinflammatory cytokines, and myeloperoxidase would have provided us more information. Moreover, we did not assess pulmonary vascular development. Because abnormal vascular development of variable severity and timing accompanies a disrupted alveolarization in BPD, information on pulmonary vascular development would have given us more comprehensive view on the pathology of BPD. These are limitations of our study [34].
In conclusion, we demonstrated that CXCR2 antagonist alleviated systemic and pulmonary inflammation induced by postnatal systemic inflammation and restored alveolarization in a newborn rat model. These results implicate that CXCR2 antagonist can be considered a potential therapeutic agent for BPD which results from disrupted alveolarization induced by inflammation.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


