Genetics of kidney development: pathogenesis of renal anomalies
Article information
Abstract
Congenital anomalies of the kidney and urinary tract (CAKUT) account for more than 50% of abdominal masses found in neonates and involve about 0.5% of all pregnancies. CAKUT has a major role in renal failure, and increasing evidence suggests that certain abnormalities predispose to the development of hypertension and cardiovascular disease in adulthood. To understand the pathogenesis of human renal anomalies, understanding the development of kidney is important. Diverse anomalies of the kidney corresponding to defects at a particular stage of development have been documented recently; however, more research is required to understand the molecular networks underlying kidney development, and such an investigation will provide a clue to the therapeutic intervention for CAKUT.
Introdction
Congenital anomalies of the kidney and urinary tract (CAKUT) account for more than 50% of the abdominal masses found in neonates and involve about 0.5% of all pregnancies1). Despite recent advances in prenatal diagnosis and early surgical intervention, these diseases still remain the primary cause of kidney failure in infants and children. Registry data from across the world, regarding the primary renal disease-mediated development of end-stage renal failure in children show that approximately 50% of the cases are due to CAKUT2, 3), and increasing evidence suggests that these abnormalities predispose children to hypertension and cardiovascular disease during adulthood4).
A wide spectrum of diseases is encompassed by the term "CAKUT," including kidney anomalies such as aplasia, hypoplasia, and multicystic dysplastic kidneys; ureteric anomalies such as megaureter, ureteropelvic junction obstruction, ureterovesical junction obstruction or incompetence, and duplex kidneys/ureters; and anomalies of the bladder and urethra5), a group that reflects the common causes of these malformations. These anomalies have a familial pattern, involving incomplete and variable penetrance, often leading to the production of different anatomical patterns. Therefore, these assorted structural anomalies have been speculated to share a common pathogenic mechanism and genetic background6). Renal functional impairment may develop postnatally from bacterial pyelonephritis and/or persistent urinary flow impairment causing renal atrophy and fibrosis, but the primary "hit" in CAKUT is clearly a developmental one, resulting in renal dysplasia5). Recently, diverse developmental anomalies of the kidney corresponding to defects at a particular stage of development have been reported (Table 1).
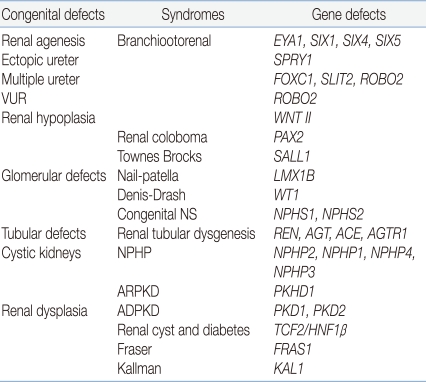
The Syndrome and Genetic Mutations Associated with Renal Tract Malfomations
Normal development of the human kidney begins at the fifth week of gestation when the ureteric bud extends from the mesonephric duct into the metanephric blastema. Development of the kidney requires complex interactions between epithelial cells in the ureteric bud and the surrounding mesenchymal cells; these interactions result in controlled cell proliferation, death, and differentiation. The ureteric bud branches serially and differentiates into the collecting ducts and ureter, with the distal end forming the bladder trigone. Meanwhile, a part of the mesenchyme undergoes epithelial conversion to form the nephron from the glomerulus to the distal tubules, with the remainder developing into the supporting renal interstitium4, 7). Ureteric bud induction and differentiation is now known to depend on the interactions mediated by growth factors, matrix molecules, and transcriptional factors that control the expression of these genes5).
Ureter induction
1. Renal agenesis
The most crucial event in kidney development is the first signaling process that induces the outgrowth of the ureter from the mesonephric duct. Central to the induction process is the expression of the signaling molecule glial cell line-derived neurotropic factor (GDNF), which is released from the mesenchyme and binds to its receptors, namely, receptor tyrosine kinase (RET) and GDNF-family receptor α1 (GFRA1), in the mesonephric duct8, 9). After binding to its receptors RET and GFRA1, GDNF activates a signaling cascade that eventually leads to epithelial cell proliferation and epithelial branching8, 10). Thus, the failure of GDNF-RET signaling is reported to be linked to renal agenesis. Renal defect in Gfrα1 knockout mice has been reported11), but mutations in GDNF or RET have not been found in human patients with kidney defects. This could be because human kidney development is not susceptible to slight reduction in protein level that results from heterozygous inactivation, and homozygous mutations can escape detection because of embryonic lethality4). However, mutations in eyes-absent homologue 1 (EYA1), sine oculis homeobox homologue 1 (SIX1), and SIX5-the 3 regulators of GDNF-result in branchiootorenal syndrome12). Outside of the metanephric mesenchyme, the signaling molecule bone morphogenetic protein 4 (BMP4) inhibits GDNF activity, and the expression of gremlin 1 (GREM1) leads to the inactivation of BMP4. Thus, mice carrying mutations in Grem 1 show renal agenesis due to Bmp4-mediated inhibition of Gdnf signaling13), and mutations in BMP4 have been reported in human renal hypodysplasia patients14).
2. Ureter malformation
Receptor tyrosine kinase antagonist sprouty 1 (SPRY1) acts downstream of GDNF by inhibiting intracellular RET signaling; mutations in this gene result in ectopic ureters by rendering the duct more sensitive to GDNF15).
Forkhead box protein C1 (FOXC1), slit homologue 2 (SLIT2), and its receptor roundabout homologue 2 (ROBO2) have been reported to confine GDNF expression to the caudal part of the nephric cord in mice studies16), and mutations in genes encoding these proteins lead to an expansion of GDNF expression to the rostral part, resulting in multiple ureters. Particularly, missense mutations of ROBO2 have been reported to be associated with vesicoureteral reflux (VUR)17, 18).
Primary VUR is a common disease, occurring with an incidence of approximately one in 100 infants, and it accounts for a high familial occurrence with a prevalence of 27-50% among siblings and offspring of patients, indicating autosomal dominant inheritance with reduced penetrance18). At present, VUR has been widely accepted as a genetically heterogeneous disorder19, 20), and genes participating in ureter induction are under investigation.
Ureteric bud branching
1. Renal hypoplasia
The ureteric bud branches serially, and nephrons are induced by a signal from the tip of the ureter. Because the kidney size is primarily determined by the total number of nephrons formed during the development, defect in the ureteric bud branching results in reduced nephron number and functional renal tissue. Recent studies have shown that there is a strong correlation between the number of nephrons and the risk of primary hypertension, with the average hypertensive patients having 46% fewer glomeruli than those in normotensive individuals21), supporting the hyperfiltration hypothesis22).
Growth of the ureter requires continuous inductive signals from the mesenchyme. WNT11, which is produced by the ureteric bud tips, seems to complete a feedback loop that acts on the mesenchyme to promote GDNF expression, and mutations that inactivate Wnt11 lead to the development of kidneys that are 36% smaller than those of the wild type23). Similarly, a reduction in GDNF-RET signaling results in reduced amounts of WNT11, and removing one copy of WNT11 and RET further aggravates the hypoplastic kidney phenotype.
GDNF-RET signaling is very crucial; mutations that affect the expression of transcriptional regulators of these genes also result in reduced ureter branching, and heterozygous mutations in paired-box protein 2 (PAX2), which activates the expression of both GDNF and RET, cause renal coloboma syndrome in humans24).
Patterning
The first glomeruli form at 8-9 weeks of gestation, and new nephrons are added at the outer layer of the cortex until 32-36 weeks. On induction, nephron precursor cells condense to form a cap structure that surrounds the ureter. These cells then undergo a mesenchyme-to-epithelial transition to form the renal vesicle, which develops into a comma-shaped body and then an S-shaped body. The S-shaped body elongates and differentiates into the various segments of a nephron. Each nephron segment fulfils particular physiological functions, such as blood filtration, pH regulation, and reabsorption and secretion of solutes. Gene expression patterns are segment specific, and defects at this stage of development result in specific glomerular and tubular diseases, but knowledge on the genetic program that leads to nephron segmentation is limited.
Podocyte cells mature under the control of transcription factors such as Wilms' tumor transcription factor (WT1). WT1 in presumptive podocyte layer induces vascular growth through the activation of vascular endothelial growth factor (VEGF) that attracts endothelial cells, which in turn produce platelet-derived growth factor (PDGF) that supports the differentiation of mesangial cells. Hence, WT1 mutation in humans is associated with diffuse mesangial sclerosis in Denys-Drash syndrome25). Terminal differentiation of the podocyte layer requires the regulation of LIM homeobox transcriptional factor 1β (LMX1B), and lack of LMX1B interferes with the expression of podocyte-specific genes, mutations in which cause nail-patella syndrome26).
CAKUT phenotype
Recently, there have been enormous advances in the field of kidney development, and genes functioning at specific developmental stages have been identified. However, expression patterns are not always restricted to a single cell lineage, and phenotypes can vary greatly depending on the type of mutation in a particular gene.
The critical role of rennin-angiotensin system (RAS) during kidney development is widely accepted, and mutations in the genes encoding components of the RAS or pharmacological inhibition of RAS results in diverse malformations of the kidney and urinary tract. Of the 2 receptors for angiotensin II, angiotensin type 2 receptor has been recently recognized, and its gene AGTR2 is primarily an embryonic gene, which is actively transcribed from the onset and throughout the embryonic development of the kidney and urinary tract system6). It has been reported that in Agtr2 mutant mice, a wide anatomical spectrum of anomalies are randomly noted in the same pedigree27), with each specific anatomical pattern having its own counterpart in humans. WTI is required for ureter induction, formation of the nephron, and for differentiation and maintenance of the glomerulus. Hence, mice carrying Wt1 mutations lack kidneys, show renal dysplasia, or develop renal failure at adulthood4). Point mutations that affect specific sites of the RET protein lead to a wide range of CAKUT phenotypes28), probably because point mutations might specifically affect the interactions of the RET protein with other regulatory proteins, leading to the development of a diverse spectrum of phenotypes. Therefore, genotype-phenotype studies of CAKUT at a large scale will be required to provide a clue for the investigation of kidney development.
Cystic kidney disease
Cystic diseases of the kidney (CDK) are a group of distinctive kidney diseases, leading to the formation of dilated tubules or cysts. They are among the most frequent genetic lethal diseases in humans. Autosomal dominant forms of CDK, the most prominent example of which is autosomal dominant polycystic kidney disease (ADPKD), are characterized by end-stage renal disease (ESRD) in adulthood. Autosomal recessive form of CDK, such as nephronophthisis (NPHP) and autosomal recessive polycystic kidney disease (ARPKD), cause ESRD in childhood or adolescence.
By positional cloning, the first 2 genes that have been implicated in CDK-polycystic kidney disease 1 and 2 (PKD1 and PKD2), which encode the proteins polycystin1 and 2, respectively-have been identified29, 30). The 2 proteins have a role in the maintenance of renal tubular cell differentiation. ARPKD is characterized by bilateral renal cystic enlargement; most patients develop ESRD in infancy, early childhood, or adolescence. By using positional cloning, researchers identified polycystic kidney and hepatic disease 1 (PKHD1), which encodes a novel protein fibrocystin/polyductin that has a role in terminal differentiation of the collecting duct and biliary systems31). NPHP is an autosomal recessive cystic kidney disease and the most frequent genetic cause for ESRD in the first 3 decades of life. Unlike PKD, cysts in ESRD are mostly restricted to the corticomedullary border of the kidneys, and kidney sizes are reduced or normal. In all, 5 NPHP-associated genes, NPHP1-NPHP5, have been identified by positional cloning; these genes encode nephrocystin proteins, nephrocystin 1-nephrocystin 5, respectively. These proteins are involved in cell-cell and cell-matrix adhesion signaling events and in cell division32). Nevertheless, the molecular link between these functions and the development of renal cysts remains elusive.
Despite their clinical and pathological heterogeneity, the similar outcome of cyst formation has led to the assumption that gene defects in each of these disorders may disrupt a common pathway. Recent studies revealed that cystoproteins, the products of cystic kidney disease genes, are expressed in the sensory organelles known as primary cilia, in the basal bodies or centrosomes33). Primary cilia link mechanosensory, visual, osmotic, gustatory, and other stimuli to the mechanisms of cell-cycle control and epithelial cell polarity. The link between the cilium and centriole suggests a mechanism underlying cilial sensing required to maintain the growth-arrested phenotype of mature tubules34).
The pathogenic link between cystoprotein expression in the cilia and the renal cystic phenotype remains unknown, and one of the CDK variants medullary cystic kidney disease 2 (MCDK2) is caused by mutations in uromodulin (UMOD); the expression of this gene has not been thus far found in the cilia, basal bodies, or centrosomes33). Nevertheless, this "ciliopathy" model for explaining the mechanisms underlying various cystic diseases in the unifying theory is an attractive hypothesis.
Renal dysplasia
Dysplastic kidneys are abnormally developed kidneys with poorly branched/differentiated nephrons and collecting ducts, increased stroma and occasional cysts, and metaplastic tissues, such as cartilage5). Although many dysplastic kidneys develop by chance, some underlying causes, including genetic defects, lower urinary tract obstruction, and teratogen/drugs, might also exist7). These conditions are not mutually exclusive and may occur simultaneously. Multicystic dysplastic kidneys (MCDK) are classically associated with non-patent ureters and obstruction, perhaps reflecting improper ureteric canalization35). Animal studies in fetal sheep and mice suggest that postnatal urinary tract obstruction mimic human dysplastic kidneys, but the size of the cysts is never as large as that in MCDK, metaplastic cartilage does not form, and there is marked inflammation in the postnatal models, suggesting that other factors besides flow impairment are implicated in MCDK7, 36), and that not all human dysplasia are associated with obstruction. Serial ultrasonography examinations before and after birth indicate that MCDK can enlarge and involute into an "aplastic" phenotype37). Involution may represent an imbalance of programmed cell death and growth by proliferation because apoptosis in human dysplasia is prominent, especially in the stroma around the dysplastic tubules38). Recent studies showed that disordered expression of diverse growth factors, cell survival genes, and transcription factor genes correlate with disordered cell turnover and maturation, resulting in excessive growth followed by apoptotic involution. Thus, renal dysplasia is considered as a dynamic disorder associated with abnormal kidney development. Upregulated proliferation occurs in human dysplastic epithelia, which may be critical for cyst formation, whereas there is excess apoptosis in the adjacent mesenchyme, which may be responsible for the adequate involution of dysplastic kidneys. These aberrant patterns of proliferation and cell death may partially be explained by abnormal expression of key nephrogenic molecules such as the transcription factor hepatocyte nuclear factor-1β (HNF-1β) and other growth factors, survival molecules, or adhesion/matrix-associated proteins.
HNF-1β, first identified as a liver-specific transcription factor, is also highly expressed in other epithelial organs, including kidneys and pancreas; in the kidney, HNF-1β is restricted to the epithelial cells comprising renal tubules and collecting ducts39). Mutations in the genes encoding HNF-1β produce an autosomal dominant disorder maturity-onset diabetes of the young, type 5 (MODY5), also known as renal cysts and diabetes (RCAD), because affected individuals present with severe cystic kidney disease40). Recently, mutations in HNF-1β have been identified in children with sporadic renal hypoplasia/dysplasia and are known to be a frequent cause of bilateral hyperechogenic kidneys detected by prenatal ultrasonography41).
Besides HNF-1β, many individual components of dysplastic kidney have been reported, such as PAX242) and uroplakins43), but some studies also report of children with compound heterozygote mutations in several renal developmental genes. Therefore, interactions between different factors need to be identified, and probably dynamic models of gene networks will have to be considered to understand these complex developmental processes.
Conclusion
In recent years, there have been enormous advances in our knowledge regarding human kidney development; however, more research is required to understand the molecular networks underlying kidney development. From an epithelial bud and a loose aggregate of mesodermal cells to a completely functional kidney, a fine tuning of network is needed, and the investigation of this networking will provide a clue to the therapeutic intervention for CAKUT.