A case of Bartter syndrome type I with atypical presentations
Article information
Abstract
Bartter syndrome (BS) is an autosomal recessively inherited rare renal tubular disorder characterized by hypokalemic metabolic alkalosis and hyperreninemic hyperaldosteronism with normal to low blood pressure due to a renal loss of sodium. Genetically, BS is classified into 5 subtypes according to the underlying genetic defects, and BS is clinically categorized into antenatal BS and classical BS according to onset age. BS type I is caused by loss-of-function mutations in the SLC12A1 gene and usually manifests as antenatal BS. This report concerns a male patient with compound heterozygous missense mutations on SLC12A1 (p.C436Y and p.L560P) and atypical clinical and laboratory features. The patient had low urinary sodium and chloride levels without definite metabolic alkalosis until the age of 32 months, which led to confusion between BS and nephrogenic diabetes insipidus (NDI). In addition, the clinical onset of the patient was far beyond the neonatal period. Genetic study eventually led to the diagnosis of BS type I. The low urinary sodium and chloride concentrations may be caused by secondary NDI, and the later onset may suggest the existence of a genotype-phenotype correlation.
In summary, BS type I may have phenotype variability including low urine sodium and chloride levels and later onset. A definitive diagnosis can be confirmed by genetic testing.
Introduction
The hallmarks of Bartter syndrome (BS) are renal salt wasting and hypokalemic metabolic alkalosis accompanied by normal or low blood pressure despite secondary hyperaldosteronism1). BS is clinically categorized into antenatal BS and classical BS2). Antenatal BS is severe, early-onset form, characterized by maternal polyhydramnios, prematurity, intrauterine and postnatal polyuria, hypercalciuria, nephrocalcinosis, normomagnesemia, recurrent vomiting, growth retardation, increased renal synthesis and urinary excretion of prostaglandin3-5). Classical BS is associated with milder phenotype with a later onset beginning from infancy to adolescence. BS is caused by genetic disruptions of ion transporters or channels in the thick ascending limb of loop of Henle6); BS type I is caused by loss-of-function mutations of SLC12A1 encoding the apical sodium-potassium-chloride cotransporter (NKCC2), BS type II by loss-of-function mutations of KCNJ1 encoding the apical inwardly-rectifying potassium channel (ROMK), BS type III by loss-of-function mutations of CLCNKB encoding the basolateral chloride channel (ClC-Kb), BS type IV by loss-of-function mutations of BSND encoding barttin, and BS type V by gain-of-function mutations of CASR encoding the basolateral calcium sensing receptor (CaSR)2). Patients with BS type I or II usually present as antenatal BS, and BS type III usually manifest as classic BS. BS type IV is specifically accompanied by sensorineural deafness, and BS type V is characterized by hypocalcemia due to hypoparathyroidism2, 7-9). The cardinal manifestation of BS is metabolic alkalosis with increased urinary electrolytes due to renal salt wasting in the thick ascending limb of loop of Henle. Typical laboratory tests typically reveal urine potassium ≥20 mEq/L, urine chloride ≥20 mEq/L, serum bicarbonate ≥30 mEq/L, blood pH ≥7.45, and increased serum renin and aldosterone levels.
Here, we report a case with BS type I with later onset of atypical manifestations mimicking nephrogenic diabetes insipidus (NDI) such as low urine sodium and chloride concentrations without definite metabolic alkalosis.
Case report
The patient was a 21-month-old male from healthy unrelated parents. He was prematurely born by elective caesarean section with a birth weight of 1,780 g at 33+4 weeks of gestation (10-50th percentile for gestational age). The pregnancy was complicated by severe polyhydramnios, requiring several amniocenteses. Immediately after birth, he was admitted to the neonatal intensive care unit of a regional medical center for 21 days. A postnatal chromosome study revealed a karyotype of 47, XYY. At the age of 1 month, he developed frequent vomiting. At the age of 7 months, his mother tried a weaning diet but failed because he could not swallow any solid foods. However, his growth was normal.
At the age of 13 months, he developed urinary tract infection. At that time, his height was 78.6 cm (50-75th percentile), weight 10.5 kg (50-75th percentile), and head circumference 46.5 cm (50-75th percentile). However, his development was retarded as 8-month-old state. In addition, suspicious bilateral medullary nephrocalcinosis were detected by renal ultrasonography.
At the age of 17 months, he was evaluated at a local hospital due to continuous vomiting. His height was 83.5 cm (75-90th percentile), weight 11.5 kg (50-75th percentile) and head circumference 47.5 cm (50-75th percentile). Development was a 9-month-old state. Brain MRI revealed slightly increased subarachnoid space, and esophagography was normal. At that time, hypokalemia was detected for the first time and kidney ultrasonography revealed definite bilateral medullary nephrocalcinosis. Thereafter, he could not grow adequately but began to lose his weight. He was referred to our hospital at the age of 21 months for further evaluation.
A thorough evaluation of the patient was done at our hospital. He did not have frequent vomiting any more, but he still had difficulty eating a solid diet. When solids were put in his mouth, he sucked out the fluids and then spat out the remainder of the food. His diet consisted of 2-2.5 L of liquid materials and urine output was 1.5-2 liters a day. His body weight was 10.8 kg (third-fifth percentile), height 89.9 cm (50-75th percentile) and head circumference 48 cm (10-25th percentile). He could walk but with unstable walking posture. He could not climb up the stairs. He could speech simple words such as mamma, papa, water and sister, but he could not speak a sentence. His blood pressure was 100/54 mmHg. Dental examination displayed generalized delayed tooth eruption state, but there were no decayed teeth or periodontal disease. Esophagography revealed slight degree of passage delay and minimal gastroesophageal reflux. Brain computed tomography showed no abnormality. Plasma renin activity was 40.8 ng/mL/hr (normal range <16 ng/mL/hr), and serum aldosterone 888 pg/mL (normal 50-194). Serum sodium level was 140 mmol/L, potassium 3 mmol/L, and total CO2 25-31 mmol/L. Serum calcium level was 10.5 mg/dL, phosphorus 5.2 mg/dL, magnesium 2.4 mEq/L, creatinine 0.5 mg/dL. Urine specific gravity was 1.003, and there was no proteinuria or hematuria. In spot urine, sodium concentration was 7 mmol/L, potassium 38.5 mmol/L, chloride 9 mmol/L, and calcium (5.0 mg/dL) to creatinine (3.3 mg/dL) ratio 1.52. Tubular reabsorption of phosphate was 82%, and trans-tubular potassium gradient was 13.78 (normal <2).
After the evaluation, BS was suspected by clinical features including hypokalemia, hyperreninemia, hyperaldosteronemia, polyuria, hypercalciuria, and nephrocalcinosis. However, low urine electrolytes levels were inconsistent with BS. Thus, for the possibility of nephrogenic diabetes insipidus (NDI), water deprivation test and genetic test were done. After 12-hour water deprivation, urine specific gravity was 1.008 and urine osmolality 276 mOsm/kg. We analyzed the causative genes of both BS and NDI, namely SLC12A1 encoding NKCC2, KCNJ1 encoding ROMK, CLCNKB encoding ClCKb, AVPR2 encoding arginine vasopressin V2 receptor and AQP2 encoding vasopressin- regulated water channel aquaporin 2 (AQP2). Compound heterozygous mutations in the SLC12A1 gene were detected; c.G1277A causing p.Cys(TGT)436Tyr(TAT) in exon 10 and c.T1679C causing p.Leu(CTT)560Pro(CCT) in exon 12. The former is a previously reported mutation in a patient with antenatal BS10), and the latter is a novel mutation. L560P mutation was not detected in 50 control Korean and 50 control Japanese. In addition, two computer programs (the Scale-Invariant Feature Transform (SIFT, http://blocks.fhcrc.org/sift/SIFT.html) and the Polymorphism Phenotyping (Polyphen, http://genetics.bwh.harvard.edu/pph/) predicted that L560P substitution would be pathogenic. There was no pathogenic mutation in other genes tested.
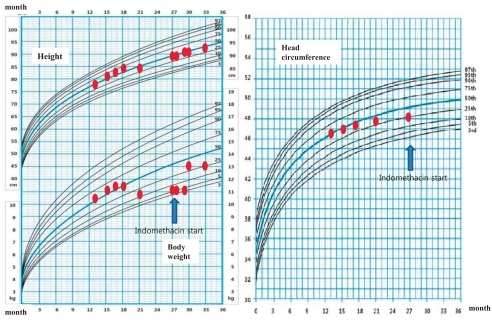
After confirming the genetic diagnosis of BS type I, indomethacin therapy was started with a dosage of 0.05 mg/kg/day, and the dosage was increased to 0.2 mg/kg/day. This regimen was well tolerated and considerably improved his clinical course. At the last follow-up at the age of 29 months, his body weight was 13.2 kg (50-75th percentile) and height was 91 cm (50-75th percentile).
Discussion
The diagnosis of BS type I in this patient was confirmed by genetic study. However, the patient had at least two atypical clinical features that are inconsistent with BS type I. In this patient, BS was clinically suspected first on the basis of typical laboratory findings including hypokalemia, hyperreninemia, hyperaldosteronemia, hypercalciuria, nephrocalcinosis as well as typical clinical features including normal or low blood pressure, polydipsia, polyuria, vomiting, maternal polyhydramnios and prematurity history. However, the urine sodium and chloride concentrations were persistently low and metabolic alkalosis was not definite (Table 1). Proesmans5) reviewed the change in the urine electrolytes levels in patients with BS during neonatal and early infant periods and found that urine sodium (93-126 mmol/L) and chloride (89-132 mmol/L) levels were high and potassium level was low (1-15 mmol/L) during the neonatal period. However, after 3-6 weeks, urine sodium concentration decreased and the initially very low urine potassium levels started to increase. This transformation of renal sodium loss to renal potassium loss may be explained by a combined maturation of both the proximal and distal tubules, which results in compensation of urinary sodium loss but at the price of marked hyperaldosteronism. However, in our patient, data about urine electrolytes during the neonatal period were unavailable and the urinary chloride level was persistently low. Thus, possibility of combined primary or secondary NDI was considered in our patient. He showed almost no response to water deprivation test and genetic study of AVPR2 and AQP2 revealed no pathogenic mutation, which suggest that he had secondary NDI. Acquired NDI can develop secondary to various causes including relief of obstruction in obstructive uropathy, hypokalemia, renal failure, lithium toxicity and hypercalcemia11). The expression of AQP2 decreases markedly in the collecting ducts of obstructed kidneys, which is consistent with the view that local factors, such as increased urine flow and pressure, can be responsible for the acquired form of NDI by affecting the AQP2 expression11, 12). Hypokalemia may cause an arginine vasopressin-resistant concentration defect11), and the reduced expression of AQP2 has been demonstrated in hypokalemia as in the other forms of acquired NDI13). In addition, a flow activated maxi-K channel and a flow-activated epithelial Na-channel (ENaC) in the distal convoluted tubules may lower urinary sodium concentration by increasing sodium influx during water diuresis2, 12, 14). In our patient, high urine output and hypokalemia may lead to the development of secondary NDI.
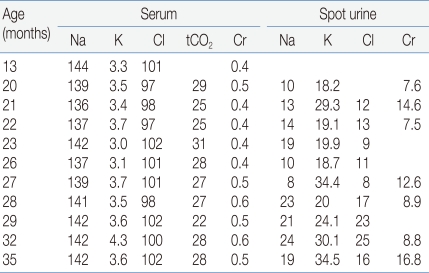
Serial Laboratory Data of the Patient
Second, although our patient had a history of prematurity with maternal polyhydramnios, the onset of symptom started far beyond the neonatal period, which was consistent with classic BS. BS type I is usually presented with antenatal or neonatal BS2). Antenatal or neonatal BS is characterized by prenatal or neonatal onset and life-threatening severe symptoms starting from early neonatal period15). There have been several case reports of patients with BS type I and atypically mild symptoms of late onset16-18). Complete loss of human NKCC2 transport activity was described for the SLC12A1 mutations associated with the severe phenotype19). Pressler et al16) suggested that the relatively mild phenotype might be explained by some residual activity of the affected NKCC2 proteins. Because one of the mutations only partially impaired NKCC2 function, variable degrees of NKCC2 dysfunction may be associated with variations of the clinical phenomenon. They described two brothers with compound heterozygous mutations in the SLC12A1 gene (p.F177Y and p.D918fs), who were diagnosed as BS at age 13 and 15, respectively. Functional study of these two mutant proteins revealed that p.F177Y mutant had significant residual transport activity. Furthermore, they performed 36Cluptake experiments after heterologous expression of wild type rat NKCC2 and mutated proteins in Xenopus oocytes. Functional analysis of the mutated rat NKCC2 protein after heterologous expression in Xenopus oocytes revealed significant residual transport activity of the NKCC2 p.F177Y mutant construct in contrast to no activity of the NKCC2-D918fs frameshift mutant construct. However, coexpression of the two mutants was not significantly different from that of NKCC2-F177Y alone or wild type. Membrane expression of NKCC2-F177Y as determined by luminometric surface quantification was not significantly different from wild-type protein, pointing to an intrinsic partial transport defect caused by the p.F177Y mutation. The partial function of NKCC2-F177Y, which was not negatively affected by NKCC2-D918fs, therefore explained a late-onset mild phenotype-associated SLC12A1 gene mutation. Yamazaki et al17) also reported a patient with BS type I (compound heterozygous for p.A555V and p.G809V mutations) who presented with asymptomatic proteinuria at age 20 years. Our patient also has compound heterozygous missense mutations (p.C436Y and p.L560P). These findings suggested that the phenotype severity may be depend on the residual transport activity of the mutant NKCC2 (genotype-phenotype correlation) in patients with BS type I. In addition, Bettinelli et al18) reported phenotypic variability in several patients with BS type I, including absence of hypokalemia and/or metabolic alkalosis in the first years of life as well as persistent metabolic acidosis mimicking distal renal tubular acidosis and presence of hypernatremia and hyperchloremia mimicking NDI.
Our patient has a karyotype of 46, XYY, a chromosomal anomaly. However, there has been no report of XYY syndrome associated with BS or NDI. XYY syndrome most often accompanies no medical problem and the syndrome is usually detected during genetic analysis for another reasons.
In summary, BS type I may have phenotype variability including low urine sodium and chloride levels and later onset, and a definitive diagnosis can be confirmed by genetic study.
Acknowledgements
This study was supported by a grant (A080588) from the Korea Healthcare technology R&D Project, Ministry for Health, Welfare and Family Affairs, Republic of Korea.