An atypical phenotype of hypokalemic periodic paralysis caused by a mutation in the sodium channel gene SCN4A
Article information
Abstract
Familial hypokalemic periodic paralysis is an autosomal-dominant channelopathy characterized by episodic muscle weakness with hypokalemia. The respiratory and cardiac muscles typically remain unaffected, but we report an atypical case of a family with hypokalemic periodic paralysis in which the affected members presented with frequent respiratory insufficiency during severe attacks. Molecular analysis revealed a heterozygous c.664 C>T transition in the sodium channel gene SCN4A, leading to an Arg222Trp mutation in the channel protein. The patients described here presented unusual clinical characteristics that included a severe respiratory phenotype, an incomplete penetrance in female carriers, and a different response to medications.
Introduction
Familial hypokalemic periodic paralysis (OMIM no. 170400) is an autosomal-dominant disorder characterized by reversible attacks of flaccid paralysis of the skeletal muscles with concomitant hypokalemia. The onset of the initial episode occurs within the second decade of life, and usually presents during adolescence. Paroxysmal muscle weakness mainly develops at night or early in the morning, and these episodes usually do not affect the respiratory and cardiac muscles. Paralytic attacks are frequently triggered by emotional stress, meals rich in carbohydrates and salt, strenuous exercise, or cold exposure1, 2).
Mutations in the CACNA1S gene (OMIM no. 114208), which encodes the alpha 1-subunit of the skeletal muscle L-type voltage-dependent calcium channel, are responsible for the majority (70%) of cases of hypokalemic periodic paralysis. Missense mutations in the SCN4A gene (OMIM no. 603967), which encodes the skeletal muscle voltage-gated sodium channel alpha subunit, account for approximately 10% of cases3, 4).
In this report, we describe a case of familial hypokalemic periodic paralysis with unusual clinical manifestations associated with a rare mutation in the SCN4A gene. This mutation results in a codon change from arginine to tryptophan at residue 222 (Arg222Trp), which is located in the critical voltage sensor of the sodium channel.
Case report
A 16-year-old boy was referred to our clinic for the evaluation of a 6-year history of episodic attacks of flaccid paralysis with intermittent respiratory insufficiency. Paralytic attacks had a variable frequency, ranging from daily to once every several months. However, an acute attack may occur after ingestion of carbohydrate- or sodium-rich meals, cold exposure, or vigorous exercise. The paralytic attacks mainly affected the hands, the feet, or both, and were often generalized and lasted for hours and sometimes days. During severe attacks, the patient experienced breathing difficulties and was put on ventilator support in a hospital several times over the past few years. His father and an uncle had similar paralytic attacks; however, the frequency and severity of the attacks had decreased in their forties, and the paralytic symptoms had occurred rarely in recent years. One of the patient's uncles had died during a severe attack in his twenties (Fig. 1).

Pedigree of the family with hypokalemic periodic paralysis. The proband is indicated with an arrow. Asymptomatic mutation carriers are shown as symbols with a split line.
At admission, there were no abnormal findings on physical examination, electrocardiography, and radiological and laboratory evaluations. Holter monitoring for 24 hours did not show structural abnormalities. During a severe attack of weakness, however, the patient developed numbness in his hands and feet, followed by a flaccid paralysis involving the entire body, with a rating of 0 using the Medical Research Council scale5). Sensory examination revealed no deficits. Deep tendon reflexes were absent in all limbs, and Babinski and Hoffmann signs were not present. Cognitive and autonomic functions were preserved. Dyspnea with a respiration rate of 40 breaths/minute developed after about 1 hour. The heart and lungs showed no abnormalities. Blood gas analysis revealed the following results: pH, 7.3 (normal, 7.35-7.45); PaCO2, 52 mmHg (normal, 35-45 mmHg); HCO3, 25 mEq/L (normal, 22-26 mEq/L), and a low serum potassium level, 1.5 mEq/L (normal, 3.5-5.0 mEq/L). Electrocardiography revealed hypokalemic findings, such as ST segment depression and prominent U waves. The patient developed cyanosis in spite of high flow oxygen delivery by a mask, and oxygen saturation was 80%, and was then put on ventilator support. Eight hours after intravenous potassium chloride infusion, blood potassium level rose to 3.5 mEq/L, and he recovered completely from the paralytic symptoms and respiratory insufficiency.
Prophylactic treatment with acetazolamide at a starting dose of 250 mg twice daily and a potassium supplement (from 1,200 mg to 1,800 mg twice daily) were administered to the patient and the other affected family members for the symptoms of episodic paralysis. This treatment was however ineffective, and they were prescribed a combination of oral spironolactone (25 mg twice daily), amiloride (5-10 mg twice daily), and potassium salts (1,200-1,800 mg twice daily). The frequency and intensity of their paralytic attacks were markedly reduced after administration of this drug combination. Serum electrolyte values, which were regularly assessed on an outpatient basis to avoid the possible risk of developing hyperkalemia, were within normal ranges throughout the combination treatment.
Mutation screening was performed by sequence analysis of the entire coding region of the CACNA1S and SCN4A genes. Genomic DNA was extracted from the peripheral blood of the patient and members of his family using a DNA extraction kit (SolGent, Daejun, Korea). All participants provided written informed consent, and the study was conducted in compliance with the Institutional Review Board of Konyang University Hospital.
Full exons of the CACNA1S and SCN4A genes were amplified to scan for mutations. Primers were designed from the introns surrounding the exons using Primer3 software (http://www.genome.wi.mit.edu/cgi-bin/primer) or VectorNTI10 (Invitrogen, Carlsbad, CA, USA). Polymerase chain reaction (PCR) was performed using a thermal cycler (GeneAmp PCR System 9700; Applied Biosystems, Foster City, CA, USA) in a total volume of 25 µL containing 100 ng of genomic DNA, 10 pmol of each primer, and 2X H-Taq Multiplex premix, which consisted of PCR buffer, deoxyribonucleoside triphosphates, MgCl2, and H-Taq DNA polymerase (SolGent). The PCR conditions were as follows: initial denaturation at 95℃ for 15 minutes, followed by 35 cycles at 95℃ for 20 seconds, at 60℃ for 20 seconds, and at 72℃ for 30 seconds, with final extension at 72℃ for 2 minutes. The PCR products were purified using a Millipore FB plate (Millipore, Billerica, MA, USA), and were then used as a template for cycle sequencing with the BigDye terminator cycle-sequencing ready reaction kit (Applied Biosystems, Foster City, CA, USA). Nucleotide sequences were determined by using an automatic genetic analyzer (model ABI 3730XL; Applied Biosystems). The obtained sequences were compared with the sequences from the control samples using SeqMan software (DNASTAR, Madison, WI, USA), and sequence variations were confirmed by analyzing both DNA strands.
Direct sequencing of SCN4A exon 5 revealed a heterozygous C>T transition at nucleotide 664 (Fig. 2), leading to the substitution of a highly conserved, positively charged arginine with a tryptophan at codon 222 (Arg222Trp). This mutation was found in the clinically affected father, uncle, and cousin, and also in the clinically unaffected grandmother and aunt (the cousin's mother) (Fig. 1). Paternity was confirmed using 15 markers provided by the PowerPlex 16 system (Promega, Madison, WI, USA).
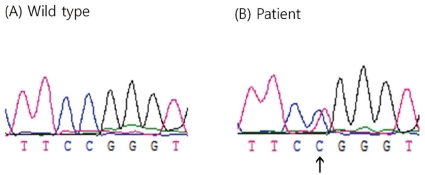
Sequence chromatograms from a normal control subject (A) and the index patient (B). A heterozygous c.664 C>T transition in exon 5 of the SCN4A gene was detected in the patient.
Discussion
Mutations in the SCN4A gene have been shown to be responsible for approximately 10% of cases of hypokalemic periodic paralysis3, 4). Among the mutations of the SCN4A gene that have been identified, the Arg222Trp is a very rare mutation that has been mentioned in only one previous study to date4). Reported here is the first case of familial hypokalemic periodic paralysis with the Arg222Trp mutation from an Asian country. We regard this mutation as the cause of clinical presentation of our patients based on the following reasons: (1) this mutation is located in the S4 segment of domain I, a functionally important part that confers voltage-sensing properties to the sodium channel, (2) the alteration of the critical S4 regions has been associated with loss of normal channel function and (3) arginine mutations in S4 segments account for 90% of hypokalemic periodic paralysis cases4). Confirmation of functional significance in a bioassay or in additional reports of affected patients with the same mutation will help to further establish the pathogenic role of the Arg222Trp mutation.
The patients described in this report possess an atypical respiratory phenotype responsible for sudden episodes of breathing difficulty. Death due to paralysis of the respiratory muscles or arrhythmic complications during attacks in hypokalemic periodic paralysis has been considered exceptionally rare2). However, the respiratory and cardiac muscles have been reported to be affected in patients with hypokalemic periodic paralysis6, 7), and the paralytic attacks described here can be potentially life-threatening. Although genotype-to-phenotype correlations have been described in hypokalemic periodic paralysis8), respiratory insufficiency has not previously been reported in association with the Arg222Trp mutation in the SCN4A gene. The presence of respiratory failure in the patient described here may be the result of ethnic differences; environmental factors; or perhaps, the Arg222Trp mutation per se. This mutation is so rare that a full description of its phenotypic correlates is yet to be reported.
Incomplete penetrance of mutations of the SCN4A gene in patients with hypokalemic periodic paralysis was reported in a Chinese family with the Arg672His mutation9). In the present case, the patient's grandmother and aunt, who had three and one genetically and clinically affected sons, respectively, had the Arg222Trp SCN4A mutation, but had never experienced paralytic symptoms. This is the first report of incomplete penetrance associated with the Arg222Trp mutation.
Acetazolamide has been considered highly effective in the treatment of patients with hypokalemic periodic paralysis10). However, in some patients with either a specific calcium channel mutation or a sodium channel mutation, no effect and even worsening of the paralytic symptoms have been reported after acetazolamide treatment3, 11). Spironolactone and its derivatives have been used as alternatives to acetazolamide in those cases in which initial empirical therapy with acetazolamide has been ineffective12, 13). We initially prescribed acetazolamide to the patients described in this report; however, the episodes of muscle weakness did not improve in either frequency or severity following acetazolamide treatment. In contrast, all patients showed significant improvements in paralytic symptoms after the administration of a combination of spironolactone, amiloride, and potassium chloride. It remains to be elucidated whether the improvements in paralytic symptoms in these patients are associated with genetic or allelic variations in response to medications.
In conclusion, the patients described in this report presented with unusual clinical manifestations of familial hypokalemic periodic paralysis, including a severe respiratory phenotype, an incomplete penetrance in women, and a different response to medication. Additional reports of patients with the same mutation would help clarify the clinical phenotype associated with this rare mutation.
Acknowledgment
This work was supported by the Korea Research Foundation Grant funded by the Korean Government (MOEHRD, Basic Research Promotion Fund) (KRF-2008-331-E00158).