Pathogenesis and clinical manifestations of juvenile rheumatoid arthritis
Article information
Abstract
Juvenile rheumatoid arthritis (JRA) is the most common rheumatic childhood disease; its onset is before 16 years of age and it persists for at least 6 weeks. JRA encompasses a heterogeneous group of diseases that is classified according to 3 major presentations: oligoarthritis, polyarthritis, and systemic onset diseases. These presentations may originate from the same or different causes that involve interaction with specific immunogenetic predispositions, and result in heterogeneous clinical manifestations. An arthritic joint exhibits cardinal signs of joint inflammation, such as swelling, pain, heat, and loss of function; any joint can be arthritic, but large joints are more frequently affected. Extra-articular manifestations include high fever, skin rash, serositis, and uveitis. The first 2 types of JRA are regarded as T helper 1 (Th1) cell-mediated inflammatory disorders, mainly based on the abundance of activated Th1 cells in the inflamed synovium and the pathogenetic role of proinflammatory cytokines that are mainly produced by Th1 cell-stimulated monocytes. In contrast, the pathogenesis of systemic onset disease differs from that of other types of JRA in several respects, including the lack of association with human leukocyte antigen type and the absence of autoantibodies or autoreactive T cells. Although the precise mechanism that leads to JRA remains unclear, proinflammatory cytokines are thought to be responsible for at least part of the clinical symptoms in all JRA types. The effectiveness of biologic therapy in blocking the action of these cytokines in JRA patients provides strong evidence that they play a fundamental role in JRA inflammation.
Introduction
Juvenile rheumatoid arthritis (JRA) is a generic term for arthritis that has an onset before the age of 16 and persists for more than 6 weeks. The JRA nomenclature represents an exclusion diagnosis that includes all forms of chronic childhood arthritis of unknown origin. JRA is the most common chronic rheumatic illness in children and is a significant cause of both short- and long-term disabilities. The heterogeneity of this disease suggests that different factors likely contribute to its pathogenesis. The current understanding of JRA indicates that it arises in a genetically susceptible individual due to environmental factors1-3). Moreover, it has been proposed that an antigen-driven autoimmune process mediates the inflammatory pathology of some cases of arthritis (e.g., oligoarthritis, polyarthritis). In contrast, there are no signs of lymphocyte-mediated, antigen-specific immune responses in individuals with systemic onset disease. Recent investigations in the pathophysiology of systemic onset disease have indicated that this disorder is due to an uncontrolled activation of the innate immune system4). Regardless of the differences in the underlying pathogenesis of the various types of JRA, proinflammatory cytokines are consistently overproduced and are related to the clinical manifestations in all types of JRA. Modulation of these cytokines results in improvement of clinical outcome5), which strongly suggests that these cytokines play important roles in JRA.
Currently, 3 separate classification systems are used to categorize individuals under 16 years of age with chronic arthritis. These include the American College of Rheumatology (ACR)6), the European League Against Rheumatism (EULAR)7), and the International League of Associations for Rheumatology (ILAR)8) criteria. Because none of these systems are perfect models, some JRA patients fulfill criteria for more than one subtype, whereas others are difficult to classify into any of the specific subgroups. Both the ACR and EULAR criteria are based solely on the onset type as it is manifested during the first 6 months of disease, whereas the ILAR criteria also include the course type over an unknown period of time thereafter, in order to further distinguish the group of patients with oligoarticular disease.
Epidemiological studies of JRA have been hampered by a lack of standardized criteria and case ascertainment, which has resulted in wide-ranging results. For instance, the reported prevalence of JRA ranges from 0.07 to 4.01 per 1,000 children, and the annual worldwide incidence varies from 0.008 to 0.226 per 1,000 children9). However, the actual incidence and prevalence of JRA in the Asian population, including Korean children, have not been well quantified because most large epidemiologic studies performed to date have been based on populations of patients who were mainly of European ancestry. Of note, one previous study from Japan showed that JRA had a relatively low overall prevalence among the population10), suggesting lower incidence and prevalence in children of Asian origin than those of European origin. Furthermore, there are significant differences in JRA subtype distribution among the different ethnic groups as well11).
Pathogenesis
1. Associations of human leukocyte antigen (HLA) and non-HLA molecules in JRA
The genetic basis of JRA is complex, but it has been estimated that the sibling recurrence risk of developing the disease is around 1512). To date, only 2 genetic risk factors, HLA and protein tyrosine phosphatase non-receptor 22 (PTPN22) genes, have been unequivocally confirmed as JRA susceptibility genes in multiple populations. The most well-established genetic factors for JRA are the HLA genes. Because the main function of HLA molecules is presenting antigenic peptides to T cells, HLA associations with JRA imply that this disease may be caused by an unidentified arthritogenic antigen. Many associations between subsets of JRA and the various HLA molecules have been described previously in the literature13-15). However, both the strength of these associations and the associated alleles vary between the JRA subtypes. Specifically, oligoarthritis has been consistently associated with HLA-A2, HLA-DRB1*11, and HLA-DRB1*08. Rheumatoid factor (RF)-positive polyarthritis is reportedly associated with HLA-DR4 in children, similarly to in adults. Moreover, the presence of HLA-B27 confers an increased risk of enthesitis-related arthritis. PTPN22 encodes a lymphoid-specific phosphatase (Lyp). A variant in the coding region of this gene, which is reportedly associated with a number of autoimmune diseases, has also been identified as a susceptibility locus for JRA16). The effect size of PTPN22 varies somewhat between JRA subtypes but, in general, is more consistent than that of HLA genes. A few other genes, including macrophage inhibitory factor, interleukin (IL)-6, IL-10, and tumor necrosis factor (TNF)-α, have also been associated with JRA in different populations and subtypes17). However, these discussed genes may collectively account for only a small proportion of the total genetic contribution to disease.
2. Inflammatory mediators of joint damage
Synovial membranes of JRA patients contain activated T and B cells, plasma cells, and activated macrophages that are recruited via an intense neovascularization process. Host tissue cells, including activated synovial fibroblasts, chondrocytes, and osteoclasts, mediate cartilage and bone destruction. It has been established that the recruitment, activation, and effector function of each of these contributor lineages are directed principally by a network of cytokines (Fig. 1).
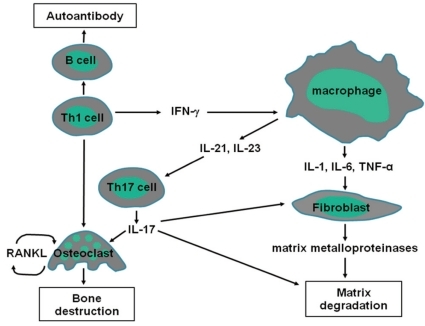
Cytokine signaling pathways involved in JRA. Interactions among macrophages, T cells, B cells, and non-hematopoietic cells including fibroblasts are important in the pathogenesis of JRA. These interactions are facilitated by the actions of cytokines that induce the production of other proinflammatory cytokines.
Antigen-specific T cells appear to play a central role in the pathogenesis of arthritis subtypes within JRA. T-cell infiltrates are composed predominantly of T helper 1 (Th1) cells, which express an activated memory phenotype and high concentrations of chemokine receptors18, 19). Th1 cells stimulate B cells, monocytes, macrophages, and synovial fibroblasts to produce immunoglobulins and inflammatory mediators. Activated B cells produce immunoglobulins, including RF and antinuclear antibodies (ANAs). The precise pathogenic role of RF remains unknown, but it may involve the activation of complement through the formation of immune complexes. ANAs, which are mainly associated with early-onset oligoarthritis, have been reported to react against different nuclear targets, none of which are specific for JRA. Activated macrophages, lymphocytes, and fibroblasts, as well as their products including vascular endothelial growth factor (VEGF) and osteopontin, can stimulate angiogenesis. VEGF is highly expressed in synovial tissue20), whereas osteopontin is raised in synovial fluid and tissue, and correlates with new vascularisation21).
TNF-α and IL-1 produced by activated monocytes, macrophages, and synovial fibroblasts likely have primary roles in the pathogenesis of JRA. These cytokines are detected in synovial fluids or tissues in a majority of JRA patients22, 23), and are known to stimulate mesenchymal cells, such as synovial fibroblasts, osteoclasts, and chondrocytes, to release tissue-destroying matrix metalloproteinases24). TNF-α and IL-1 also inhibit synovial fibroblasts from producing tissue inhibitors of metalloproteinases. Collectively, these dual actions seem to lead to joint damage. Indeed, data from animal models strongly suggest TNF-α and IL-1 play roles in JRA. For instance, transgenic mice that expressed a deregulated human TNF-α gene spontaneously developed an inflammatory and destructive polyarthritis similar to JRA25). Moreover, blocking TNF-α with either a soluble TNF-receptor fusion protein or monoclonal antibodies also ameliorated disease activity in mice with type II collagen-induced arthritis26, 27). Injection of IL-1 into the knee joints of rabbits has been demonstrated to result in the degradation of cartilage28), whereas the injection of antibodies against IL-1 ameliorated collagen-induced arthritis in mice and decreased the damage to cartilage29).
IL-6 is a multifunction cytokine that has a wide range of biological activities in various target cells and regulates immune responses, acute phase reactions, hematopoiesis, and bone metabolism30). Circulating levels of IL-6 are markedly elevated in patients with JRA, and are associated with laboratory and clinical variables of disease activity31, 32). IL-6 stimulates hepatocytes and induces the production of several acute-phase proteins, such as C-reactive protein (CRP)33). Thus, elevated levels of IL-6 in serum correlate with CRP levels in JRA patients with active disease.
IL-17 is produced by Th17 cells, and induces a massive tissue reaction due to the broad distribution of the receptors to this cytokine. Recent evidence suggests that IL-17-producing Th17 cells have a crucial role in autoimmune inflammation34). In particular, IL-17 promotes a proinflammatory cytokine environment in the joint, stimulating macrophage production of TNF-α and IL-135), and synergizes with these cytokines to increase IL-6 and IL-8 production36, 37). In addition, IL-17 contributes directly to joint destruction by upregulating matrix metalloproteinases and stimulating osteoclastogenesis through receptor activation of nuclear factor-κB ligand (RANKL) induction38-40). IL-17 is increased in JRA patients with active disease compared with levels in individuals in remission41). Data from animal models also suggest IL-17 has a role in cartilage degradation. For instance, IL-17-deficient mice were demonstrated to be resistant to induction of collagen-induced arthritis42). Moreover, joint inflammation and cartilage and bone destruction were suppressed after administration of anti-IL-17 antibodies in mice with collagen-induced arthritis43).
3. Unique inflammatory profile of systemic onset disease
The pathogenesis of systemic onset disease differs from that of other types of JRA in several respects, such as the lack of association with HLA type44) and the absence of autoantibodies and autoreactive T cells45). Thus, patients with systemic onset disease do not show signs of lymphocyte-mediated antigen-specific immune responses. Instead, the typical clinical signs of systemic onset disease are associated with granulocytosis, thrombocytosis, and upregulation of acute-phase reactants, which indicate an uncontrolled activation of the innate immune system4). During both the initial manifestation and the flares of systemic onset disease, there is a perivascular infiltration of neutrophils and monocytes producing proinflammatory cytokines involved in the pathogenesis of this disease46). The predominant role of the innate immune system in systemic onset disease is also underscored by the high expression and serum concentrations of the calcium-binding proteins S100A8, S100A9, and S100A12, which are specifically secreted during activation of neutrophilic granulocytes and monocytes47). The extraordinarily high serum concentrations of these proteins are closely associated with the disease activity of systemic onset disease, and are not found in patients with other forms of inflammatory arthritis or other autoimmune or infectious diseases48, 49). S100 proteins exhibit proinflammatory effects on leukocytes and endothelial cells, and are thus likely to be directly involved in the inflammatory process of systemic onset disease50, 51).
Recent data indicate that IL-1 has a prominent role in systemic onset disease. Treatment with IL-1 receptor antagonist has been shown to reduce the clinical and laboratory features of disease activity in patients with systemic onset disease who show resistance to anti-TNF-α treatment52). In addition, activated monocytes from patients with systemic onset disease secrete significantly higher amounts of IL-1 in comparison with secreted levels from monocytes of healthy controls, whereas release of TNF-α and IL-6 is not significantly different between these groups. Another member of the IL-1 cytokine family, IL-18, was found to be extremely elevated in patients with adult-onset Still's disease compared with levels in patients with a variety of other rheumatic diseases and in healthy controls53). An equally dramatic rise in IL-18 concentration has been found in serum from children with systemic onset disease, whereas levels in children with polyarticular or oligoarticular disease were not significantly higher than levels in the healthy controls54). Moreover, IL-18 concentrations are significantly higher in patients with either serositis or hepatosplenomegaly than in patients without these manifestations.
The circulating concentration of IL-6 is noticeably increased in patients with systemic onset disease and correlates with the extent of joint involvement55, 56). In addition, IL-6 concentration is significantly higher in the synovial fluid of patients with systemic onset disease than in patients with other JRA subtypes57). The overproduction of IL-6 may explain many of the extra-articular manifestations of this disease, including microcytic anemia58) and growth impairment59). Treatment with a monoclonal antibody directed against the IL-6 receptor is associated with pronounced clinical improvement and the reestablishment of normal levels of acute-phase reactants in patients with systemic onset disease60).
The clinical and pathologic manifestations of macrophage activation syndrome (MAS) are thought to result from the activation and uncontrolled proliferation of T-lymphocytes and well-differentiated macrophages, which leads to an unrestricted release of inflammatory cytokines, such as TNF-α, IL-1, and IL-6. However, the cause of the immunologic derangement associated with MAS is unknown. Recently, studies have revealed markedly decreased natural killer cell function and, in some cases, depressed perforin expression in patients with systemic onset disease; it has been suggested that these abnormalities may explain the distinctive susceptibility of these patients to developing MAS61, 62).
4. Anti-inflammatory mediators in JRA
The two most well-known anti-inflammatory cytokines associated with JRA are IL-10 and IL-4. IL-10 has been shown to reverse cartilage degradation mediated by antigen-stimulated mononuclear cells in adult patients with arthritis63). In addition, a single nucleotide polymorphism connected to lower production of IL-10 is associated with a more severe type of arthritis64). IL-4 inhibits the activation of Th1 cells, which in turn decreases the production of TNF-α and IL-1 and inhibits cartilage damage65). IL-4 and IL-10 cooperate to inhibit the production of inflammatory cytokines, including IL-6 and IL-866). Higher levels of IL-4 and IL-10 mRNA within a joint are allied with a milder oligoarticular course and non-erosive disease67).
Foxp3+CD4+CD25+ regulatory T cells (Tregs) are important for controlling inflammatory processes68). In humans, an X-linked genetic defect in Foxp3 is the underlying cause of a condition that presents with multiple autoimmune conditions, which is named the immuno-dysregulation, polyendocrinopathy, enteropathy (IPEX) syndrome69). Less serious defects in Treg function have also been put forward as a cause of failed tolerance in several human autoimmune diseases. However, there is currently no evidence suggesting defects in Treg function in JRA, although the number of synovial Tregs is significantly lower in patients with extended oligoarthritis compared with the number in patients with a milder course of the disease70). Moreover, a higher number of Tregs have been found within joints of JRA patients compared with the number in peripheral blood71), which indicates an enrichment of Tregs within the inflamed joints. However, it appears that high numbers of regulatory cells in the joint fail to moderate the local inflammatory process. This finding may be related to effector T cell resistance, suppression at the site of inflammation72), or the attenuation of Treg function by local dendritic cell-derived cytokines, such as IL-673).
Clinical manifestations
1. Arthritis
An arthritic joint exhibits a number of cardinal signs of inflammation, such as swelling, erythema, heat, pain, and loss of function (Fig. 2, 3). Involved joints are often warm, but are not typically erythematous. Children with arthritis may not complain of pain while at rest, but active or passive motion typically elicits pain. Joint tenderness is usually maximal at the joint line or just over the hypertrophied, inflamed synovium. Of note, bone pain or tenderness is not characteristic of JRA and may instead indicate the possibility of a malignancy involving bone. Morning stiffness and gelling following inactivity are common manifestations of joint inflammation, but young children infrequently describe these symptoms. Often, young children do not complain of pain and instead refuse to use the affected joint entirely.
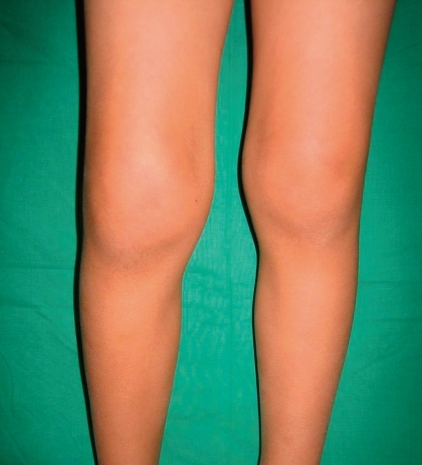
Swelling and flexion contracture of the right knee of a representative patient with oligoarticular disease.
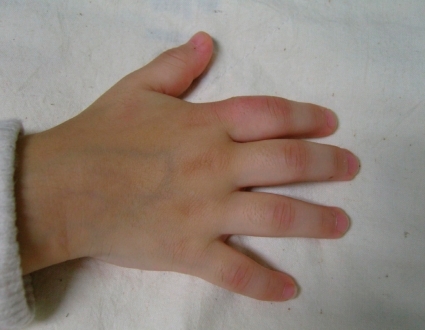
Polyarticular disease affects the joints of the wrist and hand. The proximal and distal interphalangeal joints are erythematous. There are flexion contractures of the fingers.
Any joint can be affected by JRA, but large joint are more frequently involved than smaller joints74). However, small joints of the hands and feet are also affected, particularly in polyarticular onset disease75). Of note, cricoarytenoid arthritis is unusual but may be responsible for acute airway obstruction. Inflammation of the synovial joints in the middle ear has also been detected by tympanometric studies76). The temporomandibular joint and the cervical, thoracic, and lumbar spine should also be examined in the case of JRA. JRA often affects the cervical spine, and the most common changes in the upper cervical spine are anterior atlantoaxial subluxation and impaction77, 78). Subluxation of the atlantoaxial joints may also occur, rendering the affected child at risk of injury in an accident or upon attempted intubation prior to receiving general anesthesia. Scoliosis, which possibly reflects asymmetric thoracolumbar apophyseal joint inflammation, may also occur in children with JRA. Small outpouchings of symovium are not uncommon in individuals with JRA and are particularly evident at the extensor hood of the proximal interphalangeal joint and around the wrist or ankle. A synovial cyst in the antecubital area or anterior to the shoulder may be the initial or sole presentation of JRA.
Oligoarticular disease develops in at least 50% of children with JRA during the first 6 months of disease. This subtype is the only form of JRA without an adult equivalent. Oligoarticular disease affects up to 4 joints at presentation, with the knee joints mostly affected, followed by the ankles74). In contrast, this subtype almost never affects the hips, and rarely the smaller joints of the hands and feet. Oligoarticular disease is characterized by asymmetric arthritis, early onset (before 6 years of age), female predilection, high frequency of positive ANAs, and a high risk of iridocyclitis. The ILAR classification distinguishes 2 further categories within the oligoarthritis subtype: persistent oligoarthritis, in which the disease is confined to 4 or fewer joints, and extended oligoarthritis, in which the arthritis extends to more than 4 joints after the first 6 months of disease. Up to 50% of oligoarticular patients develop extended disease, and 30% will do so in the first 2 years after diagnosis. The risk factors for extended disease include involvement of an upper limb joint and elevated erythrocyte sedimentation rate (ESR) at onset79).
Polyarticular disease is defined as the presence of arthritis in 5 or more joints during the first 6 months of disease. The arthritis may be symmetrical and usually involves the large and small joints of the hands and feet, although the axial skeleton, including the cervical spine and the temporomandibular joints, may also be affected. This subtype includes children with both RF-negative and RF-positive diseases. Both types affect girls more frequently than boys. RF-negative patients often develop polyarthritis in early childhood, whereas RF-positive patients instead develop arthritis during late childhood and adolescence. Three distinct subsets of RF-negative polyarthritis have been identified. The first subset is a form that resembles early-onset oligoarticular disease, except for the number of joints affected in the first 6 months of disease. The second subset is similar to adult-onset RF-negative rheumatoid arthritis, and is characterized by overt symmetric synovitis of large and small joints, onset during school age years, increased ESR, negative ANA, and variable outcome. Finally, the third subset is a form known as dry synovitis, which shows negligible joint swelling but stiffness and flexion contractures80). RF-positive patients are primarily adolescent girls with symmetric small joint involvement and early-onset erosive synovitis. Approximately a third of these patients develop subcutaneous nodules (non-tender, firm lesions over pressure points and tendon sheaths), typically in the board of the forearm and elbow. The HLA associations in these patients are the same as in adult seropositive rheumatoid arthritis patients and likely represent the early expression of adult rheumatoid arthritis.
2. Systemic extra-articular manifestations
Systemic involvement may precede the development of overt arthritis by weeks, months, or rarely years. In the right clinical setting, with characteristic fever and classic rash, the diagnosis of probable systemic onset disease may be made, and confirmation of the diagnosis can follow when persistent arthritis develops81). The arthritis associated with systemic onset disease is usually polyarticular affecting both large and small joints. Asymmetric, oligoarticular arthritis is less common. The systemic pattern is prominent during the first 4-6 months of disease and rarely occurs for the first time during the later course of disease.
The most prominent feature of systemic involvement is a high spiking fever82). Specifically, the temperature of an individual typically rises to 39℃ or higher on a daily or twice-daily basis, followed by a rapid return to the baseline temperature or below (Fig. 4). Although this quotidian pattern is highly suggestive of systemic onset disease, patients may not present this fever pattern. Fever may occur at any time of the day, but characteristically presents in the late afternoon to evening in conjunction with the rash. Moreover, the temperature may be subnormal in the morning. During episodes of fever, an affected child commonly appears ill when chills are present, but then appears well when the fever breaks. Fever associated with systemic onset disease often responds poorly to the commonly prescribed nonsteroidal anti-inflammatory drugs (NSAIDs), even at high doses.
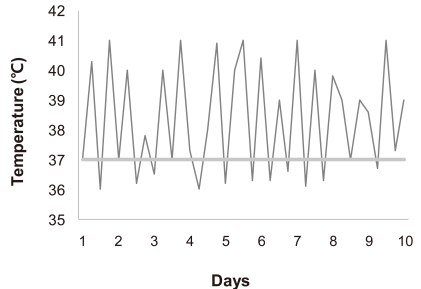
High intermittent fever in a representative patient with systemic onset disease.
In the case of systemic onset disease, intermittent fever is almost always accompanied by the classic rash82). The classic rash is evanescent (usually coming and going with the fever spikes) and consists of discrete, circumscribed, salmon-pink macules (2-mm to 10-mm in size) that may be surrounded by a ring of pallor or may develop central clearing (Fig. 5). Lesions are most common on the trunk and proximal extremities, including the axilla and inguinal areas, but can also develop on the face, palms, or soles of affected individuals. The rash tends to be migratory and is strikingly evanescent: individual lesions last for up to a few hours and leave no residua. Moreover, the rash may be much more persistent in children who are systemically very ill, and may reappear with each systemic exacerbation. Such rash also occurs very rarely in children with polyarticular onset disease, and likely never occurs in those with classic oligoarthritis. Individual lesions may be elicited either by rubbing and/or scratching the skin (the so-called Koebner response), by a hot bath, or by psychological stress. The rash is occasionally pruritic but is never purpuric.
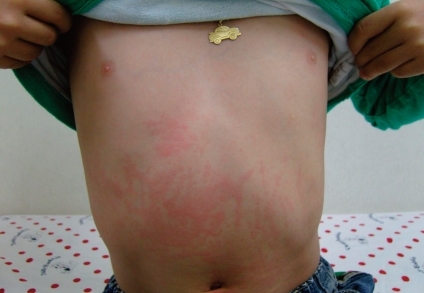
Typical rash in a patient with systemic onset disease.
Pericarditis and pericardial effusions are especially common in children with systemic onset disease83, 84). Pericarditis may precede the development of arthritis or may occur at any time during the course of disease, and is usually accompanied by a systemic exacerbation of disease. Pericarditis tends to occur in older children, but it is not related to sex, age at onset, or severity of joint disease. Most pericardial effusions are asymptomatic, although some children have dyspnea or precordial pain that may be transferred to the back, shoulder, or neck. In many cases, pericardial effusions develop insidiously, may not be accompanied by obvious cardiomegaly or electrocardiographic changes, and escape recognition except by echocardiography. Examination of affected patients may disclose diminished heart sounds, tachycardia, cardiomegaly, and a pericardial friction rub, usually at the left lower sternal border. Pneumonitis or pleural effusions may also occur with carditis, or may be asymptomatic and detected only as incidental findings on chest radiographs. Pulmonary rheumatoid nodules that are described in adult rheumatoid arthritis are rare in childhood.
Another characteristic of systemic onset disease is enlargement of lymph nodes and spleen, either alone or in combination. Marked symmetric lymphadenopathy is particularly common in the anterior cervical, axillary, and inguinal areas, and may suggest the diagnosis of lymphoma. Mesenteric lymphadenopathy may cause abdominal pain or distention and lead to an erroneous diagnosis of an acute surgical abdomen. Splenomegaly is generally most prominent within the first years after onset of systemic onset disease. The degree of splenomegaly may be extreme, but it is uncommonly associated with Felty's syndrome (splenic neutropenia)85). Hepatomegaly is less common than splenomegaly. Furthermore, moderate to severe enlargement of the liver is often associated with only mild derangement of function and relatively nonspecific histopathologic changes86). However, massive enlargement of the liver is usually accompanied by abdominal distention and pain. Progressive hepatomegaly is characteristic of secondary amyloidosis, which refers to the tissue deposition of the fibrillar protein amyloid.
MAS is a rare but life-threatening complication of systemic onset disease that is characterized by demonstration of histiophagocytosis in bone marrow87). The main manifestations of MAS include fever, hepatosplenomegaly, lymphadenopathy, severe cytopenias, serious liver disease, and disseminated intravascular coagulation. The pathognomonic feature of MAS is often found in bone marrow, which includes numerous, well-differentiated macrophages phagocytosing hematopoietic elements. It has been estimated that MAS develops in at least 10% of patients with systemic onset disease, although the true incidence of MAS might be much higher since there are no validated diagnostic criteria for this syndrome and mild instances are not always recognized88). MAS bears close resemblance to a group of histiocytic disorders collectively known as hemophagocytic lymphohistiocytosis (HLH)89). Triggers of MAS include a preceding viral illness and the addition of or a change in medications, especially including NSAIDs, intramuscular gold injections, sulfasalazine, and more recently etanercept90).
3. Uveitis
Chronic, anterior, nongranulomatous uveitis (iridocyclitis) develops in up to 21% of patients with oligoarticular disease and 10% of patients with polyarticular disease91). However, no patients with systemic onset disease have been diagnosed as having uveitis to date92). The only known independent risk factor for developing uveitis is a positive ANA test. The onset of chronic uveitis is typically insidious and often entirely asymptomatic, although up to one half of affected children have some symptoms attributable to uveitis (e.g., pain, redness, headache, photophobia, change in vision) later in the course of their disease. Uveitis may be present at the time of diagnosis, may develop during the course of JRA, or may be an initial manifestation of JRA that is usually detected in the course of routine ophthalmologic examination. JRA patients should be screened routinely to prevent delay in diagnosis of uveitis. The earliest signs of uveitis on slit-lamp examination are the presence of inflammatory cells and increased protein concentration in the aqueous humor of the anterior chamber of the eye. In addition, deposition of inflammatory cells on the inner surface of the cornea (keratopunctate deposits) may develop later during the course of disease. Complications of uveitis include posterior synechiae, cataracts, band keratopathy, glaucoma, and visual impairment.