Survival analysis of spinal muscular atrophy type I
Article information
Abstract
Purpose
The life expectancy of patients with spinal muscular atrophy (SMA) type I is generally considered to be less than 2 years. Recently, with the introduction of proactive treatments, a longer survival and an improved survival rate have been reported. In this study, we analyzed the natural courses and survival statistics of SMA type I patients and compared the clinical characteristics of the patients based on their survival periods.
Methods
We reviewed the medical records of 14 pediatric patients diagnosed with SMA type I during a 9-year period. We examined the demographic and clinical characteristics of these patients, calculated their survival probabilities, and plotted survival curves as on the censoring date, January 1, 2010. We also compared the characteristics of the patients who died before the age of 24 months (early-death, ED group) and those who survived for 24 months or longer (long-survival, LS group).
Results
The mean survival time was 22.8±2.0 months. The survival probabilities at 6 months, 12 months, 18 months, 24 months, and 30 months were 92.9%, 92.9%, 76.0%, 76.0%, and 65.1%, respectively. Birth weight was the only factor that showed a statistically significant difference between the ED and LS groups (P=0.048).
Conclusion
In this study, the survival probabilities at 2 years were far greater than expected. Because of the limited number of patients and information in this study, the contribution of improved supportive care on longer survival could not be clarified; this may be elucidated in larger cohort studies.
Introduction
Spinal muscular atrophy (SMA) is one of the most prevalent and serious single gene disorders in children. It is an autosomal recessive disorder caused by homozygous deletion of exon 7 of the survival motor neuron1 (SMN1) gene located in chromosome 5q131, 2). The incidence is 1 in 6,000-10,000 neonates3, 4). Because of the high mortality of the most serious type, the SMA type I, the prevalence, reported to be approximately 1 in 53,000 individuals, is substantially lower than that incidence5, 6).
Based on the diagnostic standards of SMA according to the European Neuromuscular Center (ENMC)7), the SMA type I (Werdnig-Hoffmann's disease) patients show overall muscular weakness and hypotonia within 6 months after birth, and could never sit by themselves. According to the previous diagnostic standards of the SMA type I8), the age at the time of death was described to be less than 2 years. But according to the standards modified in 19997), 'the lifespan of most patients is expected to be less than 2 years'. And in fact it has been reported that approximately 10% of patients developed SMA type I before 6 months of age survived even longer than 5 years9). Most SMA type I patients die because of the rapid progress of weakening of muscles and respiratory failure. But recently patients are treated with improved supportive care such as mechanical respiratory aids and sufficient nutrients supply by gastrostomy, etc., which may have beneficial influences on the survival.
Nevertheless, studies on the survival of SMA patients are not sufficient and have not yet been reported in Korea. This study aims to analyze the clinical features of the SMA type I patients with special emphases on survival statistics and to compare the characteristics of the patients who died before 24 months and those who survived up to 24 months or longer.
Materials and methods
1. Subjects
The medical records of the 14 pediatric patients satisfying the diagnostic standards for SMA type I during the 9-year period between April 2000 and April 2009 at the Severance Hospital and Gangnam Severance Hospital, Yonsei University Medical College, were reviewed. According to the diagnostic criteria of Zerre and 59th European Neuromuscular Center (ENMC) revised in 1999, SMA type I patients were defined as children who showed overall muscle weakness, hypotonia or delayed development at birth or within 6 months after birth, and who did not obtain the ability to sit down alone during the survival period. The diagnosis was confirmed by genetic analyses showing homozygous deletion of the survival motor neuron gene or by muscle biopsies.
2. Diagnostic method
SMN gene (5q12.2-q13.3) was evaluated using PCR-RFLP method. In patients who showed negative finding for exon 7 and 8 deletions, they were diagnosed with SMA type I if muscle biopsy finding showed groups of giant type I fibers mixed with fascicles of severely atrophic fibers of both histochemical types that meets diagnostic criteria of SMA type I10).
3. Statistical method
Kaplan-Meier method was used to calculate survival probabilities and plot survival curves. These analyses were conducted using death as an outcome. For patients who did not experience the respective event (death), age as of the censoring date (January 1, 2010) was used.
We compared demographic and clinical characteristics of the patients who died before 24 months and those who survived up to 24 months or longer using Mann-Whitney test for continuous variables and Fisher exact test for categorical variables. A two-sided 0.05 level of significance was used in all analyses.
Results
Among 14 patients who met inclusion criteria, 8 patients (57.1%) were male; the median gestational period was 39+0 weeks (range 33+1-40+5); the median birth weight was 3.2 kg (range 1.4-3.9); and head control was possible in 4 patients (28.6%). The median age of symptom onset was 3.0 months (range 1.0-6.0), and the median age at diagnosis was 4.0 months (range 1.0-8.0). Respiratory support including both invasive and noninvasive ventilation was given in 8 patients (57.1%). In addition, artificial feeding including L-tube feeding and gastrostomy feeding was employed in 8 patients (57.1%) (Table 1).
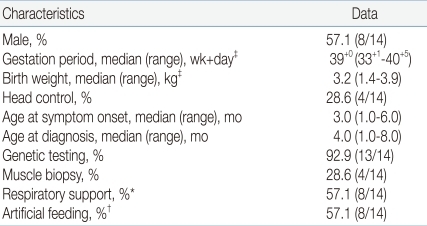
Demographic and Clinical Characteristics of Patients with Spinal Muscular Atrophy Type I
Genetic testing was performed in 13 patients (92.9%), of whom three were found to be negative for SMN1 gene deletion and subsequently diagnosed by muscle biopsy. Muscle biopsies were performed in 4 patients. The biopsy findings were round atrophic muscle fibers, diffuse atrophy with small round atrophic fiber and perimyseal fibrosis, which were compatible with SMA. In addition, two patients showed the deletion of NAIP gene. EMG was performed on 8 patients, of whom four showed the typical findings of SMA such as slow conduction velocity of motor neuron and mild to moderate denervation potential, whereas the other four showed non-specific findings.
Following the survival analysis, mean survival time was 22.8±2.0 months (Fig. 1). Survival probabilities were 92.9% at 6 months, 92.9% at 12 months, 76.0% at 18 months, 76.0% at 24 months and 65.1% at 30 months (Table 2).

Kaplan-Meier survival plot of patients with spinal muscular atrophy Type I.

Survival Probabilities (%)* at Different Ages (in Months) of Patients with Spinal Muscular Atrophy Type I
The demographic and clinical characteristics of the 10 patients (except 4 patients who were alive but did not survive more than 24 months at the censoring date) according to survival duration are summarized and analyzed (Table 3, 4). Three patients died before 24 months of age (ED) and 7 patients survived 24 months or longer (LS, 1 patient died at 26 months of age and 6 patients survived (age range: 27-79 months) until the censoring date, January 1, 2010). Four patients that survived as of the censoring date but less than 24 months did not fall into either category and were not included in the analysis. Among the 3 patients of ED group, 2 patients weighed less than 2.5 kg at birth and were small for gestational age. The other 1 patient weighed 2.8 kg. For the LS group, birth weights ranged from 2.8 to 4.0 kg in 6 patients and unknown in 1 patient. Respiratory support was employed in 2 out of 3 patients (66.7%) of ED group and 5 out of 7 patients (71.4%) of LS group. In LS group, all 5 patients that were on ventilator underwent tracheostomy. In ED group, tracheostomy was done in 1 patient, and the other patient was on non-invasive positive pressure ventilation (NIPPV). Artificial feeding was done in 66.7% of ED group and 71.4% of LS group. Among all factors, birth weight was the only factor that showed a statistically significant difference between the two groups (P=0.048).

Detailed Characteristics of 10 Patients Each of the Early-Death (ED) and Long-Survival (LS) Groups
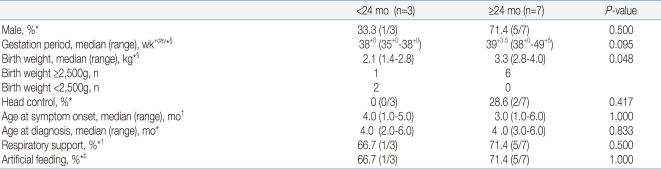
Demographic and Clinical Characteristics of Patients with Sspinal Muscular Atrophy Type I according to Duration of Survival
Discussion
SMA type I is a common, fatal, autosomal recessive disorder of childhood with an estimated incidence of 1/10,000 births and a carrier frequency of 1/50 individuals11). Major advances have been made in the past 10 years in our understanding of SMA, with identification of the SMN gene, development of animal models, and improved understanding of the SMN protein function12). Furthermore, due to intensive supportive care as mechanical ventilation and artificial feedings, the survival of patients with SMA type I has significantly increased in recent years12). Therefore, understanding the natural history and estimating survival probabilities of SMA type I patients is important not only for providing information on prognoses to parents but also for the comparison of the results of future studies that evaluate the degree of contribution of new therapeutic modalities to improvement of survival.
According to the studies reported until the early 1990s, the mean age at death of SMA type I patients was from 8.8 to 10 months, but some patients were reported to have lived up to 10 years old13-15). From the early 1990s, for SMA type I patients, noninvasive pulmonary support and gastrostomy tube feeding has been applied more widely12). In a German study reported in 1995, the survival probability at the age of 2 years was 32%, 4 years was 18%, and 10 years was 8%16). Nonetheless, reviewing the results of studies reported after the 2000s, the mean age at death was shown to be increased from 10.4 months to 4 years17, 18), and it has been reported that some patients lived up to 24 years old9). In a Hong Kong study reported in 2004, the survival probability at the age of 2 years was 40%, 4 years was 30%, and 10 years was 30%5). In addition, in a study conducted on 143 SMA type I patients reported in 2007, the survival was reported to be increased in patients born in 1995 and afterward in comparison with patients prior to 199512). In our study, the survival probability at the age of 24 months was 76.0%, which is much higher than those of previous reports described above - i.e., 32%16) and 40%5). Mean survival time calculated by Kaplan-Meier method was 22.8±2.0 months, but there may be some difficulty generalizing this data because of the statistical limitation arising from too few observed deaths at the time of censoring; that is, only 4 patients experienced the event (death).
In general, improvement of survival is thought to be mostly due to the changes in the supportive management for SMA patients5, 12, 19). In the result of a study reported in 2007, ventilatory support more than 16 hours daily, uses of mechanical insufflationexsufflation equipment, and the supply of nutrition through gastrostomy were shown to independently exert effects on survival12). Nevertheless, in a study conducted on 34 SMA type I patients in Netherland reported in 2008, the median age at death was 176 days and in regard to survival, distinct improvement was not shown, which was considered as the reflection of the difference of the attitude on medical treatments for SMA type I patients1). In our study, birth weight was the only factor that showed a statistically significant difference between the patients who died before 24 months and those who survived 24 months or longer. Due to the limitation of this study, we may not be able to state that a lower birth weight is associated with poor prognosis such as earlier deaths. However, a special attention may be given to the fact that two of the three infants of ED group were small for gestational age, whereas none of the LS group was. Implementation of respiratory support and artificial feeding was not significantly different between the two groups. This may be due to a small number of patients in the analysis. In addition, our study has methodological limitations because we could not compare the patient group with the control group who refused treatment or who were treated prior to the development of proactive treatment. Another point to consider is that whether implementation of supportive care was a prophylactic measure or a rescue. Since our study is a retrospective analysis, it is impossible to differentiate between the two.
In SMA type I paients, the known prognostic factor is the number of SMN 2 copies, and it has been shown that the more of SMN2 copies, the milder is the disease20-22). In addition, it has been reported that the disease severity of SMA could be predicted by the combination of the number of SMN2 copies and the deletion of NAIP23). SMN1 and SMN2 show 99% homology with the difference of only 5 nucleotides24). SMN1 produces full length SMN transcripts and stable SMN proteins, on the other hand, SMN2 produces the transcript lacking the exon 7 and truncated unstable SMN proteins25, 26). Nevertheless, approximately 10% SMN2 copies produce full length transcripts, and thus the increase of the number of SMN2 copies is beneficial to SMA patients, and thus exerts effects on the severity of SMA22, 27). In our study, the number of SMN2 copies in the patients was not examined, so the association of the number of SMN2 copies with survival could not be assessed. In addition, in our study, 3 patients did not show the homozygous deletion of SMN1 in genetic tests but diagnosed by muscle biopsy. Based on the result of a previous large study conducted on 525 typical SMA patients that the homozygous absence of SMN1 was identified in 92%, point mutations in 3.4%, and no mutation in 4.6%1), it is possible that the patients had a point mutation although rare or developed a disease in a different gene that is not linked to chromosome 5q13. In point mutation cases, at least one SMN1 is deleted in one chromosome, and thus to detect the presence of point mutation, a quantitative test that can determine the number of SMN1 copies may be of help1, 22). In our study, 2 patients out of 14 showed NAIP deletion, however we can not make sure if there is certain relationship with disease severity and this result was maybe due to limited number of patients.
Until now, a curative treatment for SMA is not known. It is a disease that only palliative treatments could be given. Our study has limitations that only a small number of patients were involved in the retrospective analyses, and that only death and survival probabilities were considered without detailed analysis on the quality of life of patients such as continuous ventilation and muscle strength. However, such studies on the survival are helpful to provide basic information for future evaluation of new treatments.