Perivascular epithelioid cell tumor (PEComa) of the ascending colon: the implication of IFN-α2b treatment
Article information
Abstract
A 7-year-old boy presented with hematochezia and abdominal pain. A 3.7-cm-sized mass was identified in the ascending colon by abdominal computed tomography and colonoscopy. The patient underwent surgical resection. Pathological examination revealed a low-grade perivascular epithelioid cell tumor (PEComa). PEComa in the colon is very rare. Only a few cases have been reported so far. An effective treatment method for this rare tumor has not been established yet. The patient received adjuvant interferon-α immunotherapy for 1 year. He has been tumor-free for 26 months since the initial diagnosis. This report is the first documented case of the use of interferon-α for pediatric PEComa of the colon.
Introduction
Perivascular epithelioid cell tumor (PEComa) is a family of related mesenchymal neoplasms that include angiomyolipoma, lymphangiomyomatosis and clear cell 'sugar' tumor of the lung, and a group of rare, morphologically and immunophenotypically similar lesions arising at a variety of visceral and soft tissue sites1). It has distinct histological and immunohistochemical features defined by coexpression of melanocytic (HMB-45) and smooth muscle markers (smooth muscle actin and desmin), but negativity to S-100 protein2). Data on gastrointestinal (GI) PEComa remain limited to isolated case reports. To our knowledge, 17 cases have been reported in the literature, among which 10 occurred in adults3). Because of the rarity of PEComa, its clinical, phenotypic and biological characteristics are not fully understood, and little is known about prognosis and treatment modalities for PEComa. The mainstream treatment is surgical excision. However, chemotherapy and radiotherapy have been used in locally advanced, metastatic and histologically malignant cases. We report a child with PEComa in the ascending colon, treated by surgical excision and interferon-α2b (IFN-α2b) immunotherapy.
Case report
A previously healthy 7-year-old boy was admitted to our hospital because of hematochezia and abdominal pain. There was no history of bleeding disorders, recent infection, change in bowel habits, or significant weight loss. Family history was not specific. Physical examination revealed tenderness in the periumbilical area. On digital rectal exam, there was some gross blood mixed with soft stool. A complete blood count showed white blood cell 11,500/mm3, hemoglobin 8.2 g/dL, hematocrit 26.0%, mean corpuscular volume 72.4 fL, platelet 401,000/mm3 and other biochemical tests were normal. A Meckel scan was negative. Abdominal computed tomography (CT) (Fig. 1A) and colonoscopy displayed a 3.9 cm-sized pedunculated mass at the hepatic flexure of the ascending colon. Surgical resection and right hemicolectomy was performed. Examination of the resected colon revealed a 4×3 cm in size, polypoid, yellow and partially brown mass (Fig. 1B). The tumor cells were predominantly epithelioid cells having prominent nuclei and abundant cytoplasm (Fig. 2A), and they were positive for HMB-45 (Fig. 2B), but negative for S-100 protein, actin, and desmin. The diagnosis of intestinal PEComa was established. The tumor was low-grade malignancy as evidenced by infiltrative growth but mild cellular atypia, low mitotic and proliferation index without necrosis and no vascular invasion. All of the surgical margins were free of tumor. A metastatic workup including a bone scan, positron emission tomography, brain and chest CT was negative. He had no history of primary melanoma, and no stigmata of tuberous sclerosis. He was treated with adjuvant IFN-α2b therapy (Intron-A®, Schering-Plough, Kenilworth, New Jersey, New Jersey, USA): 3 million IU/m2, 3 times/week, subcutaneous injection, for 1 year. Short term side effects, such as fever, neutropenia and skin necrosis, were not seen in the present case. He has been free of disease under regular surveillance 26 months after initial diagnosis.
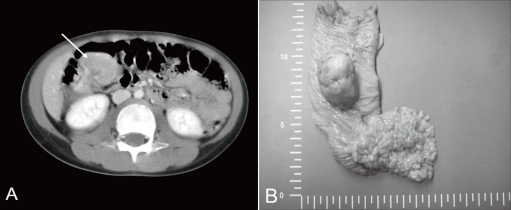
(A) Computed tomography of the abdomen showing a 3.9-cm pedunculated polyp in the right ascending colon. (B) Gross finding showing a 4×3 cm, yellow and brown, pedunculated mass at the hepatic flexure of the ascending colon. Multiple polyps in the small bowel were also visible.
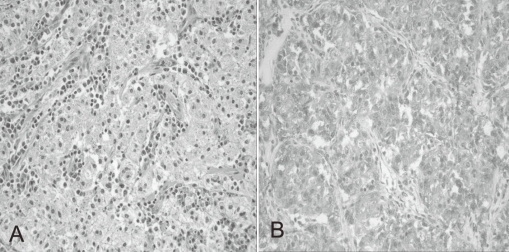
(A) Histopathological appearance of the tumor consisting of nests of epithelioid cells with eosinophilic granular cytoplasm. The nuclei of tumor cells were small and round to oval in shape (Hematoxylin & Eosin staining; ×200). (B) Immunohistochemical features of the tumor showing expression of HMB-45 antigen (HMB-45; ×200).
Discussion
A recently defined PEComa is a rare mesenchymal neoplasm of uncertain malignant potential affecting various organs. At first, Bonetti et al4) proposed the concept of perivascular epithelioid cell (PEC) in 1992 and the terminology 'PEComa' was suggested by Zamboni et al5) in 1996. The World Health Organization accepted the designation 'PEComas,' defining them as mesenchymal neoplasms that share the same distinctive cell type, the PEC in 2002.
PEComas have been reported in several organs including the uterus, falciform ligament, prostate, small and large bowel, rectum, lung, breast, cardiac septum, liver, kidney, pancreas and soft tissue of the thigh. A review of 51 cases of PEComa documented in the literature revealed that 90% of cases affected females and 41% involved the uterus6). In the pediatric population, there have been only a few documented PEComas in several organs including the duodenum, appendix, transverse colon, sigmoid colon, rectum, kidney, broad ligament, vagina and orbit. To our knowledge, 17 cases of GI PEComas have been reported in the literature, among which 10 occurred in adults3). Pediatric GI PEComas were documented in rectum3, 7, 8), appendix9), duodenum10), sigmoid colon11) and transverse colon12). This report is the eighth documented case of the pediatric GI PEComa, and the third documented case of the pediatric colon PEComa.
PEComa has distinctive morphologic, immunohistochemical, ultrastructural and genetic features. The tumor cells have large epithelioid appearance with abundant clear to granular eosinophilic cytoplasm and a large vesicular nucleus. No normal cellular counterpart has been recognized for these cells. Immunohistochemically, PEComa expresses myogenic and melanocytic markers, such as HMB45, HMSA-1, MelanA/Mart1, microophthalmia transcription factor (Mitf), actin and, less commonly, desmin13). On ultrastructural analysis, PEComa contains microfilament bundles with electron-dense condensation, numerous mitochondria and membrane-bound dense granules.
Little information is available on the possible molecular alterations occurring in PEComas. In one report, the loss of chromosome X and a t(3;10) translocation were identified by conventional cytogenetic techniques14). In another report, no gain or loss of chromosomal material was shown by comparative genomic hybridization6). Chromosomal study was not granted for this case.
Histologic criteria indicative of malignant potential in PEComas have not been established, especially in the pediatric age group. However, in a recent study of PEComa of soft tissue and gynecologic origin, Folpe et al15) suggested criteria to separate PEComas into benign, uncertain malignant potential, and malignant categories. They proposed that malignancy was predicted by the presence of 2 or more of the followings: tumor size greater than 5 cm, infiltrative tumor border, high nuclear grade and cellularity; more than 1 mitosis/50 high-power fields, tumor necrosis, and vascular invasion. In the present case his tumor was regarded as low grade, as evidenced by infiltrative tumor border but lacking of other findings listed above.
Optimal treatment for PEComas needs to be established. Primary excision is usually curative, as most tumors are benign. However, locally advanced or metastatic disease portends a poor prognosis and strategies incorporating chemotherapy, radiation and immunotherapy have been reported. Various responses, including complete, partial or absent responses, have been noted for dacarbazine, vincristine and imatinib mesylate. Rigby et al16) reported an 11-year-old girl with metastatic renal PEComa treated with dacarbazine based chemotherapy and imatinib mesylate. She presented with two large abdominal masses in the left flank and epigastrium and left supraclavicular lymphadenopathy. The tumor did not respond to an initial treatment of chemotherapy, including dacarbazine, BiCNU and vincristine. A trial of imatinib mesylate based on the expression of c-KIT was unsuccessful because of the lack of response and adverse effects of the drug.
Among 7 pediatric GI PEComa cases reported so far, six were treated with surgery alone7-12), and the last was treated with surgery and adjuvant chemotherapy consisting of doxorubicin, ifosfamide, and mesna on the basis of non-rhabdomyosarcoma soft tissue sarcoma protocol ARST0332 by Children's Oncology Group3).
Adjuvant IFN-α2b was used in patients with melanoma17), and for immunotherapy in a patient with PEComa of the bladder18). IFN-α2b was also used for other solid tumors. It reduced neomicrovascular density in the normal urothelium adjacent to the tumor after transurethral resection of superficial bladder carcinoma19). Outpatient treatment with subcutaneous IFN-α2b plus interleukin-2 treatment for metastatic renal cell cancer was also reported. Eight of 47 patients (17%) responded to this regimen and overall response rate was similar to that seen with high-dose interleukin-2 alone, however, the response duration appeared to be shorter20). While IFN-α2b therapy for the management of PEComa remains experimental, we decide to use IFN-α2b based on the vascular nature of this tumor and its anti-angiogenic effect.
The present case represents the first documented pediatric GI PEComa treated with surgical resection and adjuvant IFN-α2b immunotherapy. Further studies involving larger number of cases and close monitoring by long-term follow-up are required in the light of its potential for local recurrence and distant metastasis.