Characteristics of late-onset epilepsy and EEG findings in children with autism spectrum disorders
Article information
Abstract
Purpose
To investigate the clinical characteristics of late-onset epilepsy combined with autism spectrum disorder (ASD), and the relationship between certain types of electroencephalography (EEG) abnormalities in ASD and associated neuropsychological problems.
Methods
Thirty patients diagnosed with ASD in early childhood and later developed clinical seizures were reviewed retrospectively. First, the clinical characteristics, language and behavioral regression, and EEG findings of these late-onset epilepsy patients with ASD were investigated. The patients were then classified into 2 groups according to the severity of the EEG abnormalities in the background rhythm and paroxysmal discharges. In the severe group, EEG showed persistent asymmetry, slow and disorganized background rhythms, and continuous sharp and slow waves during slow sleep (CSWS).
Results
Between the two groups, there was no statistically significant difference in mean age (P=0.259), age of epilepsy diagnosis (P=0.237), associated family history (P=0.074), and positive abnormal magnetic resonance image (MRI) findings (P=0.084). The severe EEG group tended to have more neuropsychological problems (P=0.074). The severe group statistically showed more electrographic seizures in EEG (P=0.000). Rett syndrome was correlated with more severe EEG abnormalities (P=0.002). Although formal cognitive function tests were not performed, the parents reported an improvement in neuropsychological function on the follow up checkup according to a parent's questionnaire.
Conclusion
Although some ASD patients with late-onset epilepsy showed severe EEG abnormalities, including CSWS, they generally showed an improvement in EEG and clinical symptoms in the long-term follow up. In addition, severe EEG abnormalities tended to be related to the neuropsychological function.
Introduction
Autism spectrum disorders (ASD) are neurological and developmental disorders characterized by impairment of social relatedness, communication skills, restriction of interests and stereotyped behaviors1). ASD is believed to be related with central nervous system (CNS) dysfunction, and epilepsy is strongly associated. Epilepsy is reported to occur in one third of ASD patients but the exact prevalence is unknown with the literature reporting a wide range of estimates from 52) to 46%3). In general population, epilepsy is reported in only 0.5-1%. The fact that the epilepsy rate is much higher in ASD patients suggests that ASD and epilepsy might share a common pathophysiological basis. This could also be supported by the reports of high rates of epilepsy or epileptiform electrocencephalography (EEG) in children with autism, and vice versa, as well as much higher prevalence of autism in children with epilepsy who also have learning problems. It is unclear if this suspicion is just an epiphenomenon of an underlying neural dysfunction in ASD or there is any causal relationship between ASD and epilepsy. Many authors have not reached a definite conclusion and there is no consensus on the neuropathophysiology of ASD.
In approximately 1/3 of ASD patients, the regression of language is observed between 18-24 months of age with the appearance of autistic behavior. In childhood disintegrative disorder (CDD), autistic regression occurs after 3 years of age, and mainly includes motor regression and a loss of bowel and bladder use. According to Kurita et al.4), EEG abnormalities are significantly more common in CDD patients than infantile autism patients. However, according to Canitano et al.5), an abnormal EEG is found usually after regression but there is no evidence of a causal relationship between them. Therefore, there is still controversy regarding the role of epilepsy or epileptiform EEGs in the regression of ASD patients. Currently, there has not been any controlled study examining the efficacy of treating epileptiform EEG discharges in ASD patients. The aim of this study was to examine the clinical and electroencephlography (EEG) characteristics of autistic spectrum disorder patients who later showed clinical seizures. An attempt was made to clarify the relationship between regression or any type of problematic clinical findings and severity of EEG abnormalities in ASD patients with late-onset epilepsy, by reviewing the serial EEG findings and underlying abnormalities of the background rhythms and paroxysmal discharges.
Materials and methods
1. Subjects and methods
Thirty patients diagnosed with ASD according to Diagnostic and Statistical manual of Mental disorders of the American Psychiatric Association 4th edition (DSM-IV)6) criteria in early childhood in National Health Insurance Cooperation (NHIC) Ilsan Hospital and later developed clinical seizures, from January 2004 to May 2010, were enrolled in this study. A retrospective chart review was done, and every patient underwent more than one routine awake and sleep 30-minute-EEG. If one patient underwent several EEGs, the worst result was selected and the background rhythm and paroxysmal discharges were reviewed. The patients were divided into 2 groups according to the severity of underlying EEG abnormalities. In the severe group, Group II, EEG showed persistent asymmetry, slow and disorganized background rhythms and continuous sharp and slow waves during slow sleep (CSWS). In the other group, Group I, EEG showed a mild degree of diffuse encephalopathy and/or occasional sharp waves, and normal EEG results were also included.
2. Statistical analysis
Statistical analysis was performed by SPSS 12.0 (SPSS, Inc., Chicago, IL) for windows. A P value≤0.05 was considered significant. An independent samples T-test and one-way ANOVA were used to analyze the results.
Results
1. Patient characteristics
The subjects were 30 patients diagnosed with ASD according to DSM-IV criteria in early childhood in NHIC Ilsan Hospital and later developed clinical seizures, from January 2004 to May 2009. The age range of the patients was 4-21 years (mean 13.5 ± 4.5 years). Among them, 17 patients (56.7%) were male and 13 (43.3%) were female. There were 2 (6.7%) patients with Asperger's syndrome, 5 (16.7%) with pervasive developmental disorder not otherwise specified (PDD-NOS), 15 (50%) with Autistic disorder but no childhood disintegrative disorder (CDD), and 8 (26.6%) with Rett syndrome (Fig. 1).
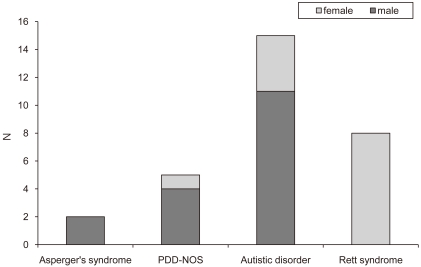
Subtype of austism spectrum disorder in 30 patients. Abbreviation: PDD-NOS, Pervasive developmental disorder, not otherwise specified.
The mean age of the onset of epilepsy in patients with Asperger's syndrome, PDD-NOS, autistic disorder and Rett syndrome was 7.5 years (range 7.1-8.0 years), 10.0±6.0 years (range 2.0-16.0 years), 7.7±5.1 years (range 2.0-21.0 years), and 4.0±3.0 years (range 2.2-10.1 years), respectively. And each group was using 1.0, 1.0, 1.5 and 2.6 anti-epileptic drugs, respectively (Table 1).
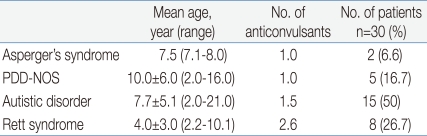
Onset Age of Epilepsy and the Number of Anticonvulsants according to the Autism Spectrum Disorder Subtypes
2. Seizure type of the patients with ASD
Among the 30 patients, 14 (46.7%) patients showed only partial seizures, 4 (13.3%) showed partial seizures with secondary generalization, and 12 (40%) showed generalized seizures.
3. Associated neuropsychological problems of the patients with ASD
Many patients suffered associated neuropsychological problems. Some suffered from several problems simultaneously and others did not suffer any problem. According to the data, 16 (53.5%, 14 versus 2) had behavioral problems, such as aggressiveness, irritability and self mutilation, 7 (23.3%, 4 versus 3) suffered mood disorders, such as depression, 5 (16.5%, 2 versus 3) had sleep disturbances, 5 (16.5%, 5 versus 0) had attention-deficit hyperactivity disorder (ADHD) or hyperactivity, 4 (13.3%, 2 versus 2) showed regression of language, 3 (10%, 3 versus 0) were treated for Tic disorders, 2 (6.6%, 1 versus 1) suffered anxiety disorders and 1 (3.3%, 1 versus 0) suffered elimination disorders, such as enuresis or encopresis, and 1 (3.3%, 1 versus 0) patient reported somatoform disorders (Table 2).
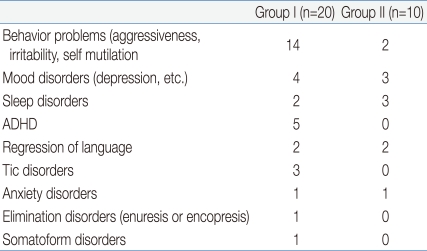
Associated Neuropsychological Problems of the Patients with Autism Spectrum Disorder
4. Family history of the patients with ASD
Many patients had associated family history. Some of them had several things simultaneously. Although 20 (66.7%) patients had no definite associated family history, 4 (13.3%) patients had a family history of epilepsy, 3 (10%) had a family history of mental retardation, 3 (10%) had a family history of manic-depressive disorder or speech delay, and 1 (3.3%) patient even had a family history of autism spectrum disorder.
5. Findings of EEG
All 30 patients were initially diagnosed with ASD in early childhood, and then with epilepsy later. Serialized routine awake and sleep 30-minute-electroencephlography (EEG) were performed after they showed clinical seizures, which was repeated until recently, even though some of them did not report clinical seizure events anymore. In average, routine EEG was performed for 5.6±3.8 times, (range 1-16 times), and follow up period was 4.9±1.9 years (range 1.3-9.0 years).
To analyze the result, the worst result from serialized EEGs was selected and the background rhythms and paroxysmal discharges were reviewed. Thirteen (43.3%) showed a normal background rhythm, whereas 7 (23.3%) showed a slow background rhythm for age, 1 (3.3%) showed inconsistent asymmetry, 2 (6.6%) showed persistent asymmetry, 4 (13.2%) showed a poverty of the normal background rhythm, and 3 (10%) patients showed slow and disorganized background rhythm.
Regarding the paroxysmal discharges, 7 (23.3%) patients showed no definite epileptiform discharges, 15 (50%) showed focal spikes or sharps, 2 (6.6%) showed generalized sharp and wave discharges (GSW), and 6 (20%) exhibited continuous sharp and slow waves during slow sleep (CSWS) (Table 3). Overall, there were 20 and 10 patients in groups I and II, respectively.
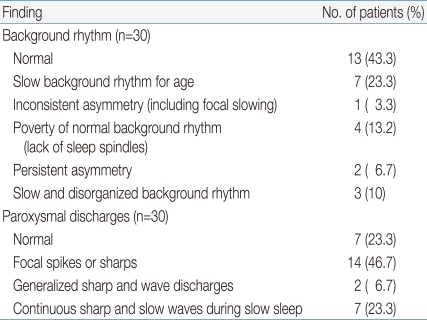
Electroencephalography Findings
6. Clinical findings according to the EEG severity group
Table 4 lists the clinical characteristics of the two groups. Group I included patients who showed normal to mild abnormal EEG results. Group II included patients who showed severe abnormal EEG results. Group I consisted of 15 males and 5 females, while group II was comprised of 2 males and 8 females. The mean age of groups I and II was 14.6±4.6 years and 11.1±3.5 years, respectively, showing no statistically significant difference (P=0.259). The age of the epilepsy diagnosis was also not significantly different, 8.5±4.6 years and 3.7±2.9 years in groups I and II, respectively (P=0.237). In group I, 8 patients showed generalized type of seizures and 12 showed partial seizures. In group II, 4 patients showed generalized seizures and 6 showed partial seizures. In group I, there were 2 Asperger's syndrome patients, 4 PDD-NOS, 13 Autistic disorder patients, and 1 Rett syndrome patient. In group II, there was no Asperger's syndrome patient, 1 with PDD-NOS, 2 with Autistic disorder, and 7 with Rett syndrome. There was a statistically significant difference in the patient distribution between the two groups (P=0.002). This means that the more severe type of ASD patients showed a more severe type of EEG abnormalities. The number of anti-epileptic drugs currently used was 1.4±0.8 in group I and 2.3±0.9 in group II. Group II appeared to use more anti-epileptic drugs but, there was no significant difference (P=0.493). In the selected EEG, group I and II showed 0.1±0.3 and 0.5±0.5 electrographic seizures, respectively, and group II showed significantly more electrographic seizures (P=0.000). However, there were no significant differences in family history (7/20 versus 2/10, P=0.074) or abnormal magnetic resonance image (MRI) findings (3/20 versus 3/10, P=0.084) between the two groups. Although there was no significant difference, group II patients showed greater association with neuropsychological problems (P=0.074).
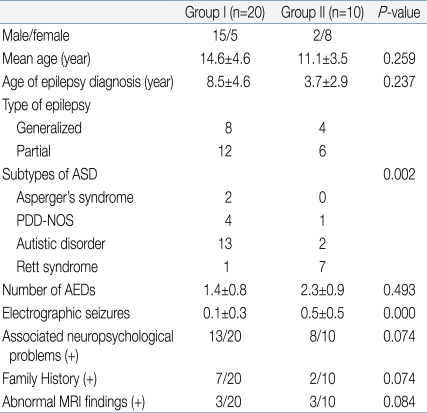
Clinical Findings according to the Electroencephalography Severity Group
7. EEG findings and clinical progress of group II
Table 4 lists the EEG findings and clinical progress of the patients in group II. There were 10 patients and of which 8 (80%) had a variety of associated neuropsychological problems. The follow up EEG improved in 8 (80%) patients after an average of 26.7±13.9 months (range 16-53 months), whereas 2 (20%) still showed continuous sharp and slow waves during slow sleep (CSWS). Seven out of eight patients who showed an improvement of EEG became seizure free, and 1 out of 2 patients who showed no improvement of EEG was seizure free. The other 2 patients, one who showed an improvement on the follow up EEG and one who still showed CSWS, suffered clinical seizure events until recently.
Discussion
According to this study, 1/3 of ASD patients with late-onset epilepsy showed definite epileptiform paroxysmal discharges or a slow and disorganized background rhythm that might affect the cortical dysfunction. Those patients largely showed associated clinical problems, such as mood disorders, sleep disturbances, behavioral problems and a regression of language. On the follow-up, 80% of them (8 out of 10) showed EEG improvement after an average of 26.7±13.9 months (range 16-53 months), and 70% (7 out of 10) showed clinical seizure improvement. The result suggests that although the EEG abnormality was severe, it was treated well by AEDs in ASD.
Autism spectrum disorders (ASD) are neurological and developmental disorders characterized by an impairment of social relatedness, communication skills, restriction of interests and stereotyped behaviors1). ASD is believed to be related with central nervous system (CNS) dysfunction, and epilepsy is strongly associated, with a wide range of estimates from 52) to 46%3). This high rate of epilepsy suggests that ASD and epilepsy might share a common pathophysiological basis. However, there is no consensus regarding the neuropathophysiology of ASD7).
According to DSM-IV6), ASD consists of five different subgroups. Asperger's syndrome is autism without mental retardation or delayed language development, with a reported epilepsy rate of 5-10% of early childhood8). PDD-NOS includes patients with generally milder autism, that does not fit criteria for any other subtypes. Autistic disorder is classic autism with all three behavioral domains showing early unexplained regression before three years of age, maximally between eighteen to twenty-four months of age9). Clinical epilepsy occurs in more than one third of children until adolescence. Childhood disintegrative disorder indicates more severe autism, and epilepsy has been reported in almost 70% of patients10). Rett's syndrome is characterized by a specific X-linked genetic deficit, mutations of the MeCP2 gene. In this group, the epilepsy rate is reported to be more than 90%11), which is the highest of all subgroups.
Many factors affect the variable rate of reported epilepsy rate in ASD. According to Tuchman and Rapin12), there is a bimodal age distribution of seizure in ASD. The first peak is in infancy before 5 years of age, and the second peak is in adolescence, after 10 years of age11). In our study, the mean age for the onset of epilepsy was 7.1±4.9 years and there was no significant difference between the ASD subtypes. This is not precisely concordant with the reported studies but the patients selected in this study could be considered as a late-onset epilepsy group.
Epileptiform discharges are rare (1-4%) in healthy children13, 14). Until recently, the reported rates of epileptiform discharges varied from 6-30% of ASD patients12, 13, 16, 17). In this study, although all patients suffered clinical seizure events, the EEG results were variable and 23 out of 30 patients (76.6%) showed epileptiform EEG abnormalities. These EEG changes might be considered a biomarker of a cortical dysfunction in this population. There are no specific epileptiform EEG patterns known in ASD patients yet. These EEG changes can show the following: slowing, asymmetry, spikes or sharps, sharp and slow waves, generalized sharp and slow waves or generalized polyspikes in diffuse or generalized, multifocal or focal discharges, unilateral or bilateral and localized to many different brain areas3, 5, 18-21). All seizure types can be associated with autism, which is consistent with our results. There is no report on the prevalence of CSWS pattern in ASD, and in this study 7 out of 30 patients (23.3%) showed CSWS.
Approximately, one third of ASD patients show a regression of language between 18-24 months of age, with the appearance of autistic behavior12). Many studies focused on the potential association between epilepsy and autistic regression. Some reported higher rates of epilepsy in children with autism and regression but others showed no association between autism, epilepsy and regression. According to Tuchman and Rapin12), about 20% of children with autistic regression, even without epilepsy, have an abnormal EEG, and Canitano et al.5) reported that this abnormal EEG occurs after regression but still there is no evidence of a causal relationship between the epileptiform abnormality and regression. In this study, we could not confirm that each patient clearly showed autistic regression but many of them had associated neuropsychological problems, which was considered to indicate a tendency to regress. Although there was no statistical significance, there was slightly higher association with neuropsychological problems in group II, which included CSWS. These results suggest that if CSWS is accompanied in late-onset epilepsy in ASD, it might cause epileptic encephalopathy, as in Landau-Kleffner syndrome, and may be a reason for the neuropsychological problems including cognitive impairment. There is no report that clarify the relationship between CSWS and autistic regression. According to Nickels et al.22), CSWS is considered distinct epileptic encephalopathy with common clinical features including seizures, regression, and epileptiform abnormalities that are activated by sleep. It is thought that CSWS is related with regression of global skills and can show behavioral phenotype that overlaps with autism23). In opposite direction, this can suggest that somehow autism and regression have possible relationship with CSWS. As most ASD patients already have impaired communication skills, this study could not confirm whether there was language regression. However, in Group II, even Rett syndrome patients showed unusual regression as well as emotional and behavioral disturbances. Overall, the relationship between the occurrence of regression and epileptiform EEG abnormalities is unclear.
Although epileptiform EEG abnormalities are much more common in children with ASD, whether we should treat these abnormalities even if the patient does not show clinical seizure is a controversial problem. Generally, interictal discharges are thought to interfere with normal neural processing. In addition, attentions are paid to the frequent cognitive and behavioral disturbances in many epilepsy syndromes24). For example, in Benign Epilepsy with Centro-Temporal Spikes (BECTS), the academic problems related to the frequency of epileptiform discharges and attention problems largely improved after EEG had normalized25, 26). In many cases of Landau-Kleffner Syndrome (LKS), both the seizures and language impairment improve with normalization of the EEG1). According to these reports, the treatment of EEG abnormalities might have a positive effect on the symptomatic improvement of children with ASD and epileptiform EEGs. Many studies reported specific improvements in ASD symptoms after therapy to suppress the discharges with AEDs27-29).
Even with these promising reports, it is difficult to make a decision as to whether to treat the epileptiform EEG abnormalities if an autistic child with no clinical seizure events has a history of regression.
In this study, the patients experienced clinical seizures some time after the diagnosis of ASD. Some of them showed nothing remarkable in the EEG but others showed definitely severe epileptiform paroxysmal discharges, such as CSWS or a slow and disorganized background rhythm that might affect the cortical dysfunction. In addition, those patients largely showed associated neuropsychological problems, which were considered to be a marker of regression or disintegration of the patient's cortical function. As every patient showed a definite clinical seizure, they were treated with anti-epileptic drugs. The follow-up revealed, 18 out of 30 patients (60%) to be seizure free with only 1 patient showing seizure aggravation. Therefore, late-onset epilepsy in ASD patients appears to be treated well by AEDs. Even in the severe EEG group, the EEG results and clinical progress improved. However, as formal cognitive function tests were not performed in all patients, and as matched untreated children were not included, it cannot be concluded that the treatment of EEG abnormality was related to the improved neuropsychological symptoms. In addition, these improvements might be due to the natural history of the disorder itself. However, these results suggest that there is some support for such an assumption.
As this study was a retrospective study and included a small number of patients at one institute, these patients might not reflect all autistic children. Therefore, a prospective, placebo-controlled, double-blinded study will be needed to further test this hypothesis.
Despite the limitations of this study, these results suggest an early intervention for seizure control when an autistic patient shows clinical seizure or neuropsychological problems. In this case, EEG should be performed to confirm if there are any epileptiform EEG abnormalities. It is natural to treat EEG abnormalities with clinical seizures. However, although there is still some controversy regarding the treatment of epileptiform EEGs without clinical seizures, as it is believed that a large portion of these ASD patients already have intellectual disability, as they show clinically problematic findings, such as seizures and maladjusted behavior without their own expressions, it appears that more should be done to improve their quality of life by screening for any type of epileptiform EEGs that can affect their brain function and treating them if possible.