Renal transplantation in a patient with Bartter syndrome and glomerulosclerosis
Article information
Abstract
Bartter syndrome (BS) is a clinically and genetically heterogeneous inherited renal tube disorder characterized by renal salt wasting, hypokalemic metabolic alkalosis and normotensive hyperreninemic hyperaldosteronism. There have been several case reports of BS complicated by focal segmental glomerulosclerosis (FSGS). Here, we have reported the case of a BS patient who developed FSGS and subsequent end-stage renal disease (ESRD) and provided a brief literature review. The patient presented with classic BS at 3 months of age and developed proteinuria at 7 years. Renal biopsy performed at 11 years of age revealed a FSGS perihilar variant. Hemodialysis was initiated at 11 years of age, and kidney transplantation was performed at 16 years of age. The post-transplantation course has been uneventful for more than 3 years with complete disappearance of BS without the recurrence of FSGS. Genetic study revealed a homozygous p.Trp(TGG)610Stop(TGA) mutation in the CLCNKB gene. In summary, BS may be complicated by secondary FSGS due to the adaptive response to chronic salt-losing nephropathy, and FSGS may progress to ESRD in some patients. Renal transplantation in patients with BS and ESRD results in complete remission of BS.
Introduction
Bartter syndrome (BS) is a rare inherited renal tubular disorder characterized by renal salt wasting, hypokalemic metabolic alkalosis and normotensive hyperreninemic hyperaldosteronism1, 2). Although BS is a typical tubular disorder, there have been several case reports of patients who developed focal segmental glomerulosclerosis (FSGS), a glomerular lesion, during the course of BS3-7). Such cases support a possible link between the two diseases, and indicate that stimulation of the renin-angiotensin system (RAS) and increased angiotensin II in response to chronic glomerular hyperfiltration due to salt-losing tubulopathy may lead to secondary FSGS3, 4, 8).
In addition to FSGS, several other factors have been reported to influence the renal survival of patients with BS, including recurrent episodes of dehydration during early infancy, chronic hypokalemia, long-term use of non-steroidal anti-inf lammatory drugs, nephrocalcinosis, and mutations in the BSND gene9-11). However, there have been only a few case reports of patients with BS who underwent renal transplantation6, 12-15).
Here, we present a case of a patient with BS who developed FSGS and subsequently underwent renal transplantation due to aggravation of his renal function. We also review previously reported cases of both diseases and cases of individuals with BS who underwent renal transplantation.
Case report
The male patient was born at term with a birth weight of 2.8 kg. There were no perinatal problems including polyhydramnios, and the family history was unremarkable. At age 3 months, the patient visited a hospital due to recurrent febrile episodes and poor weight gain. The patient was diagnosed as BS, and potassium supplement and indomethacin medication were started. However, his compliance to the drug was very poor. Detailed clinical and laboratory data from that time are unavailable.
At age 7, proteinuria was incidentally detected upon routine follow-up urinalysis. A renal ultrasonography revealed diffusely increased parenchymal echogenicity without definite evidence of nephrocalcinosis or nephrolithiasis. At age 8, a renal biopsy was conducted at the hospital due to persistent proteinuria (24-hour urine protein excretion 3,840 mg/day, serum albumin 3.4 g/dL, and serum creatinine 1.2 mg/dL). A biopsy revealed a perihilar type of segmental sclerosis in two of seven glomeruli. His proteinuria did not respond to 4 weeks of oral prednisolone (2 mg/kg/day) treatment.
At age 10, the patient was referred to our hospital due to progressive azotemia. During this time, he was taking oral potassium, ibuprofen and enalapril. The patient's height was 123cm (<3rd percentile), weight was 36 kg (90-97th percentile), and blood pressure was 117/69 mmHg at the time of admission. The laboratory examination revealed a serum creatinine level of 3.1 mg/dL, a sodium level of 138 mmol/L, a potassium level of 3.7 mmol/L, a chloride level of 103 mmol/L, a bicarbonate level of 30 mEq/L, a magnesium level of 2.0 mEq/L, a plasma renin activity of 83 ng/mL/hr (normal range 1-2.5), an aldosterone level of 558 pg/mL (normal range 50-194), and a spot urine calcium (mg) to creatinine (mg) ratio of 0.02. Additionally, the patient's bone age was 6 years 5 months upon admission. Genetic analysis revealed a homozygous p.Trp(TGG)610Stop(TGA) mutation in exon 16 on the CLCNKB gene. His renal function deteriorated progressively for one year thereafter, at which point hemodialysis was started. Subsequently, the patient underwent renal transplantation successfully from a deceased donor at age 16. The post-transplantation course has been uneventful for more than 3 years, with complete disappearance of BS without recurrence of FSGS.
Discussion
BS is a heterogeneous disorder both clinically and genetically. Clinically, BS can be classified into two variants, antenatal/neonatal BS and classic BS according to the onset age. Genetically, BS can be classified into at least 5 subtypes according to underlying mutant genes, all of which are expressed in the tubular epithelial cells of the thick ascending limb of the loop of Henle. BS type I is caused by loss-of-function mutations of SLC12A1 encoding the apical sodium-potassium-chloride cotransporter (NKCC2), while BS type II is caused by loss-of-function mutations of KCNJ1 encoding the apical inwardly-rectifying potassium channel (ROMK), BS type III is caused by loss-of-function mutations of CLCNKB encoding the basolateral chloride channel (ClC-Kb), BS type IV is caused by loss-of-function mutations of BSND encoding barttin, and BS type V is caused by gain-of-function mutations of CASR encoding the basolateral calcium sensing receptor (CaSR)2). Our patient presented with classic BS and CLCNKB mutations.
BS is an inherited tubular disorder; however, our patient developed FSGS, an acquired glomerular lesion, later in the disease course. There have been five similar case reports of BS complicated by FSGS3-7) (Table 1). In addition, there is an additional report of a patient with classic BS who developed end-stage renal disease due to glomerulopathy12). However, this patient was excluded from the review because the renal pathology was described as 'diffuse' glomerulosclerosis. As shown in Table 1, patients with BS and FSGS show development of FSGS independently of gender, clinical variants of BS or genetic subtypes of BS. One patient had nephrocalcinosis, and three patients had a history of long-term non-steroidal anti-inflammatory drug treatment prior to the onset of proteinuria. FSGS was detected during childhood to young adult age in all six cases. Three patients had progressed to chronic renal failure, but the rate of progression was variable. One other patient revealed mildly decreased renal function one year after diagnosis with FSGS5). Normal renal function was maintained in the remaining two patients, and, interestingly, proteinuria resolved completely in one of these patients after one year treatment with potassium supplements, spironolactone, and indomethacin3). The renal pathologic findings were described in detail in three cases, two of which had perihilar type FSGS. The perihilar variant is common in patients with secondary forms of FSGS mediated by glomerular hyperfiltration or other adaptation after loss of renal mass8, 16). These findings suggest that FSGS complicated by BS may be a secondary lesion due to adaptive response to chronic hyperfiltration by chronic salt-loss and resultant chronic stimulation of the renin-angiotensin system. Several case reports of Gitelman syndrome complicated by FSGS support this explanation17, 18).
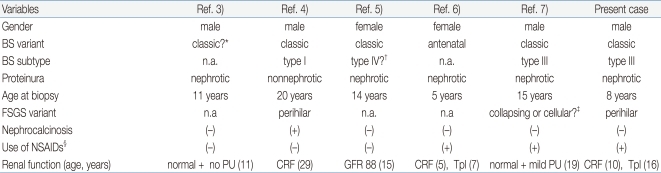
Summary of Case Reports of Bartter Syndrome (BS) Complicated by Focal Segmental Glomerulosclerosis (FSGS)
Case reports of patients with BS who underwent renal transplantation are also rare, and the causes of ESRD include progression of FSGS or other causes including complication of long-term non-steroidal anti-inflammatory drug treatment6, 12-15). In one of the patients, pre-emptive bilateral native nephrectomies and renal transplantation were conducted prior to the onset of ESRD due to severe, clinically brittle, neonatal BS15). In all of the cases, transplantation was successful and BS disappeared completely after transplantation as shown in our patient.
In summary, BS may be complicated by FSGS due to adaptive response to chronic salt-losing nephropathy, and FSGS may cause ESRD in some patients. Renal transplantation in patients with BS with ESRD results in complete remission of BS.
Acknowledgment
This study was supported by a grant (A080588) from the Korea Healthcare technology R&D Project, Ministry for Health, Welfare and Family Affairs, Republic of Korea.