Systematic review of the clinical and genetic aspects of Prader-Willi syndrome
Article information
Abstract
Prader-Willi syndrome (PWS) is a complex multisystem genetic disorder that is caused by the lack of expression of paternally inherited imprinted genes on chromosome 15q11-q13. This syndrome has a characteristic phenotype including severe neonatal hypotonia, early-onset hyperphagia, development of morbid obesity, short stature, hypogonadism, learning disabilities, behavioral problems, and psychiatric problems. PWS is an example of a genetic condition caused by genomic imprinting. It can occur via 3 main mechanisms that lead to the absence of expression of paternally inherited genes in the 15q11.2-q13 region: paternal microdeletion, maternal uniparental disomy, and an imprinting defect. Over 99% of PWS cases can be diagnosed using DNA methylation analysis. Early diagnosis of PWS is important for effective long-term management. Growth hormone (GH) treatment improves the growth, physical phenotype, and body composition of patients with PWS. In recent years, GH treatment in infants has been shown to have beneficial effects on the growth and neurological development of patients diagnosed during infancy. There is a clear need for an integrated multidisciplinary approach to facilitate early diagnosis and optimize management to improve quality of life, prevent complications, and prolong life expectancy in patients with PWS.
Introduction
Prader-Willi syndrome (PWS; OMIM 176270) is a complex multisystem genetic disorder that occurs due to the lack of expression of paternally inherited imprinted genes on chromosome 15q11-q131, 2). The syndrome has a characteristic phenotype including severe neonatal hypotonia, early onset of hyperphagia, development of morbid obesity, short stature, hypogonadism, learning disabilities, behavioral problems, and psychiatric problems3, 4). Whereas earlier prevalence estimates in the United States were in the range of 1 in 8,000-20,000, recent epidemiological surveys in Europe and Australia have estimated the lower limit of birth incidence at approximately 1 in 30,000 and population prevalence at approximately 1 in 50,0005-8). Recent surveys have highlighted the high rates and varied causes (respiratory infection or insufficiency, unexplained sudden death, choking on food, and central adrenal insufficiency) of morbidity and mortality throughout the natural course of the disease9-11). There is a clear need for an integrated multidisciplinary approach to facilitate early diagnosis and optimize management to improve quality of life, prevent complications, and prolong life expectancy in patients with PWS.
Clinical manifestations
1. Characteristics of PWS in the newborn and infant period
Severe hypotonia is consistently observed at birth and during the neonatal period (Fig. 1)12). PWS should thus be considered in all cases of neonatal hypotonia. Neonatologists appear to be on the frontline of early diagnosis including PWS in the differential diagnosis of severe neonatal hypotonia13). The first stage of PWS, occurring in infancy, is characterized by lethargy, marked hypotonia, global developmental delay, and small genitalia with frequent cryptorchidism14). Although poor weight gain is typically noted on standard infant growth charts, studies have documented excessive body fat in infants with PWS by skinfold measurements15), dual energy-x-ray absorptiometry, and doubly labeled water16). Conversely, lean body mass measurements are decreased in PWS infants, correlating with a 30% lower energy expenditure as compared to healthy individuals16).
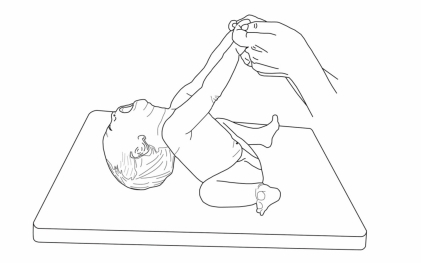
Hypotonia of infancy in patients with PWS. Note the head-lag position in traction.
2. Dysmorphic features
Patients with PWS have characteristic dysmorphic features including a narrow bifrontal diameter, almond-shaped palpebral fissures, narrow nasal bridge, and thin upper lip with a down-turned mouth (Fig. 2). These patients very commonly have shorter total hand size, narrow palms with hypoplastic hypothenar bulges, and short feet with short toes. Fair hair and hypopigmentation of the eyes and skin relative to other family members are frequently observed in patients with deletion-type PWS; these features are less commonly observed in patients with uniparental disomy.

Characteristic facial features of patients with PWS.
3. Developmental and cognitive delays
Gross motor and language milestones are delayed. Early milestones are reached on average at double the normal age (e.g., sitting at 12 months, walking at 24 months, and words at 2 years). Cognitive disability is evident by school age. Most patients with PWS are mildly mentally retarded (mean intelligence quotient (IQ): 60-70 score), with approximately 40% having borderline mental retardation or low-normal intelligence and approximately 20% having moderate retardation17-19). Impaired physical function in PWS patients during childhood is most often related to body composition abnormalities and hypotonia, rather than to linear growth deficits.
4. Hyperphagia and obesity
Obesity typically begins between 1 and 4 years of age. During later childhood, a seemingly insatiable appetite (hyperphagia) begins. This hyperphagia is hypothalamic in origin, caused by a lack of sense of satiety20, 21). Food seeking is common, and if intake is not controlled externally, central obesity results from these behaviors combined with a low metabolic rate and decreased activity level (that result in a decreased total caloric requirement). Body composition studies demonstrated both increased body fat and reduced muscle mass in patients with PWS from infancy to adulthood.
5. Sleep abnormalities
Individuals with PWS have sleep-disordered breathing, including central and obstructive sleep apnea, abnormal arousal, abnormal circadian rhythms in rapid eye movement (REM) sleep, reduced REM latency, and abnormal response to hypercapnia as well as excessive daytime sleepiness22). Obesity can worsen the sleep disorder.
6. Behavioral and psychiatric disturbances
A characteristic behavioral pattern begins in early childhood in 70-90% of affected individuals. It is typified by temper tantrums, stubbornness, controlling and manipulative behavior, compulsive-like behaviors (repeated organizing, writing, collecting, and need to finish 1 thing before moving to the next), and difficulty with changes in routine17). Autism spectrum disorder, attention deficit/hyperactivity symptoms, and insistence on sameness are common and of early onset23, 24).
7. Endocrinological abnormalities
1) Obesity
Obesity in PWS is the major cause of morbidity and mortality: cardiorespiratory failure, cor pulmonale exacerbated by obstructive and central apnea, septicemia due to skin infections, and pneumonia9, 11). Appropriate consultation with cardiologists and pneumonologists in cases of severely obese individuals is essential. A number of reports suggest that glucose tolerance is abnormal in individuals with PWS. Fasting plasma insulin concentrations and insulin response to glucose are often increased in affected individuals, suggesting insulin resistance. Type 2 diabetes mellitus has been reported in approximately 25% of adults with PWS with a mean age of onset of approximately 20 years of age25); it appears logical to approach diabetes management including weight loss and increased exercise by using similar pharmacological agents as those used for non-PWS obesity-related diabetes.
2) Growth deficiency
Short stature is usually observed, especially during the second year, because of growth hormone (GH) insufficiency exacerbated by the lack of a pubertal growth spurt. The serum levels of IGF-I are reduced in majority of children with PWS. Spontaneous GH secretion is reduced, and the GH peak during pharmacological stimulation testing is less than 10 ng/mL in 70% of children26). Adults with PWS have lower stimulated GH secretion than obese controls, but the precise prevalence of severe GH deficiency is unclear because reference ranges are unavailable for severe obesity. The mean spontaneous adult height has been reported as 162 cm in men27) and 149 cm in women28) in German cohorts. GH treatment in children with PWS improves growth during childhood, adult height, and body composition.
3) Hypogonadism
Hypogonadism is a consistent feature in both men and women with PWS; this is manifested as genital hypoplasia throughout life, incomplete pubertal development, and infertility in the vast majority of cases. In men, the penis may be small, but the most characteristic feature is a hypoplastic scrotum that is small, poorly rugated, and poorly pigmented. Unilateral or bilateral cryptorchidism is present in 80-90% of affected men. In women, the labia (majora and minora) and clitoris are generally hypoplastic. Most individuals will have no, delayed, and/or incomplete puberty. Isolated premature pubarche (probably due to early maturation of the zona reticularis of the adrenal gland) and precocious puberty have been reported in 14% and 4% of males and females, respectively29, 30). Hypogonadism is of hypothalamic origin, and generally, there is hypogonadotropism with decreased testosterone or estrogen and decreased follicle-stimulating hormone and luteinizing hormone levels in both genders. There is no consensus on the management of these conditions.
Molecular and genetic basis of PWS
PWS is an example of a genomic disorder, as it results from alterations in the structure of the genome (an epigenetic phenomenon) and not a specific change in the DNA sequence31). The various genomic changes causing PWS lead to the loss of expression of the paternally expressed genes on chromosome 15q11.2-q13 through loss or failure of expression; this is because the maternal contribution has been programmed by epigenetic factors (e.g., DNA methylation) to be silenced32). PWS and Angelman syndrome are the first known examples of human diseases involving imprinted genes33).
1. Structure and genes in the 15q11-q13 region
The 15q11.2-q13 region is highly complex and contains a number of imprinted and non-imprinted genes (Fig. 3). The majority of patients with deletions have 1 of 2 common proximal breakpoints (BP1 and BP2) and a common distal breakpoint (BP3)34-36). The paternally expressed genes are located in the more centromeric part of the region. These are MKRN3; MAGEL2; NDN; C15orf2; SNURF-SNRPN (typically referred to as SNRPN); and the C/D box small nucleolar RNA (snoRNA) genes SNORD107 (previously HBII-436), SNORD64 (previously HBII-13), SNORD108 (previously HBII-437), SNORD109A (previously HBII-438A), SNORD116 (previously HBII-85), SNORD115 (previously HBII-52), and SNORD109B (previously HBII-438B). SNORD115 and SNORD116 are present as multicopy gene clusters, whereas the other snoRNA genes are single-copy genes. In contrast to other C/D box snoRNAs, which are usually involved in the modification of ribosomal RNAs, these snoRNAs do not have a region complementary to ribosomal RNA and might be involved in the modification of mRNAs, probably by modulating alternative splicing37-39). Paternal-only expression of MKRN3, NDN, and SNRPN is regulated by parent-of-origin-specific DNA methylation of the promoter regions of each gene. Whereas the active paternal allele is unmethylated, the inactive maternal allele is methylated.
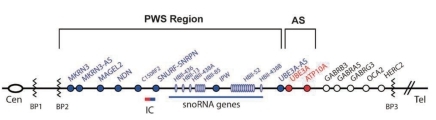
Summary of the genetic and expression map of chromosomal region 15q11.2-q13. There are 6 paternal-only (PWS region) expressed unique copy genes (MKRN3, MAGEL2, NECDIN, C15ORF2, and SNRPN and a family of 5 paternal-only expressed snoRNA genes). Only UBE3A and ATP10A, related to Angelman syndrome (AS), have maternal-only expression in mice and humans. The bipartite imprinting center lies proximal to SNRPN and within the 3-Mb PWS/AS imprinted region. The cluster of GABA receptor genes (GABRB3, GABRA5, and GABRG3), OCA2 (type II albinism), and HERC2 are not imprinted and have biparental expression. Type 1 deletions extend from BP1 to BP3, and type 2 deletions extend from BP2 to BP3 (Cassidy et al. 2009 Eur J Hum Genet17;3-13).
The most complex gene in the 15q11-q13 region is SNRPN. The original gene, found to consist of 10 exons, encodes 2 different proteins. Exons 1-3 encode SNURF, a small polypeptide of unknown function40), while exons 4-10 encode SmN, a spliceosomal protein involved in mRNA splicing in the brain41). Many 5' and 3' exons of SNRPN have been identified. These exons have 2 peculiar features: They do not have any protein coding potential, and they occur in many different splice forms of the primary transcript33). According to recent reports, the SNORD116 locus is probably a major gene contributing to the PWS phenotype42-44). It is likely, however, that 1 or more additional genes in the region also contribute to the phenotype.
1) Microdeletions of the chromosome region 15q11-q13
A 5-7-Mb de novo deletion of the proximal region of the paternal chromosome 15 [del(15)(q11-q13)], which includes the entire imprinted domain plus several non-imprinted genes, is found in the majority (~70%) of patients with PWS. At the molecular level, usually 2 classes of deletions (class I and II) can be distinguished, 1 spanning from BP1 to BP3 and the other from BP2 to BP3 (Fig. 3). A precise localization of the deletion breakpoints and the determination of the deletion sizes have been performed by array-CGH analysis recently45-47).
2) Uniparental disomy of chromosome 15
The second most common genetic abnormality in PWS (~25-30%) is a maternal uniparental disomy of chromosome 15 [upd(15) mat], occurring mostly due to maternal meiotic nondisjunction followed by mitotic loss of the paternal chromosome 15 after fertilization31). Upd(15)mat leads to the lack of expression of imprinted genes that are active on the paternal chromosome only.
3) Imprinting defect
Disruption of the imprinting process on the paternally inherited chromosome 15 is the third molecular mechanism underlying PWS (Fig. 4). This disruption is present in approximately 2-5% of individuals. Most imprinting defects are epigenetic (epimutations) and demonstrate a maternal-only DNA methylation pattern despite the presence of both parental alleles (biparental). DNA sequence changes are not found in these epimutations; they are thought to be random stochastic errors occurring during spermatogenesis in the fathers48). By contrast, approximately 15% of individuals with an imprinting defect are found to have a very small deletion in the PWS imprinting center region located at the 5' end of the SNRPN gene.
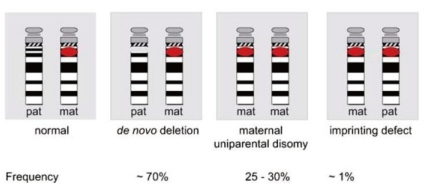
Molecular classes of PWS and their frequencies (Buiting et al. 2010. Am J Med Genet Part C Semin Med Genet 154C:365-376).
Diagnostic testing
Parent-of-origin-specific DNA methylation can be used to confirm the clinical diagnosis of PWS patients in all 3 molecular classes (deletion of 15q11-q13, uniparental disomy, and imprinting defect). The most widely used DNA methylation test targets the 5' end of the SNRPN locus49, 50). The promoter region of SNRPN is unmethylated on the paternally expressed allele and methylated on the maternally repressed allele. Normal individuals have both a methylated and an unmethylated allele, whereas individuals with PWS have only the maternally methylated allele. DNA methylation cannot distinguish this molecular class. Deletion of the paternally contributed 15q11.2-q13 is typically diagnosed using the SNRPN fluorescence in situ hybridization probe32). Chromosome analysis should be included in testing for a deletion, as occasionally the deletion is the result of a chromosomal translocation. Maternal uniparental disomy is diagnosed using DNA polymorphism analysis of the proband and parental DNA.
Treatment
Patients with PWS frequently require medical care. Initial management of hypotonia or poor feeding, evaluation for hypogonadism or hypopituitarism, management of obesity, monitoring for scoliosis, and therapy for behavioral issues are needed51). Currently, no medications have been found to effectively modify hyperphagia. Sex steroid supplementation does improve secondary sex characteristics but may aggravate behavioral disorders52).
1. GH therapy in patients with PWS
On June 20, 2000, the US Food and Drug Administration approved the use of GH in children with genetically confirmed PWS and the evidence of growth failure53). GH deficiency has been well documented in PWS by GH response testing with decreased GH response, relatively low IGF-1 levels, and abnormal body composition with high fat mass and low lean body mass. Numerous beneficial effects of GH therapy have been documented, including improvement in linear growth, physical appearance, functional muscle mass, and infant neurodevelopment. However, GH therapy does not appear to enhance the development of scoliosis, and it anecdotally modulates behavior in some patients52-54). Treatment of adult PWS patients with GH is under investigation and appears to be promising55). Currently, the recommended dose of GH is 24 U/m2 per week (1.0 mg/(m2 · day) for patients over 2 years of age. Korean insurance covers patients diagnosed with PWS by clinical symptoms and genetic testing; Genotropin (somatotropin (rDNA origin) for injection, Pfizer) is used for treatment. The optional dose of GH covered by Korean insurance is 0.035 mg/(kg · day) or 1 mg/(m2 · day) (but less than 2.7 mg/day). Treatment is covered from over 2 years of age, until the bone age is 14-15 years in girls and 15-16 years in boys. Once a female's height reaches 150 cm or a male's height reaches 160 cm, insurance coverage is no longer provided in Korea. Then, after a GH stimulation test, if the patient has GH deficiency, the patient can continue GH treatment with the adult dosage (2 U/day), which is covered by insurance.
2. GH therapy in infants with PWS
Although GH therapy in PWS has various beneficial effects, body composition and physical function (muscle strength and agility) remain abnormal even after 4 years of GH therapy, with high doses given the last 2 years56). These persistent manifestations of the syndrome might reflect non-GH-related abnormalities intrinsic to PWS and/or the late initiation of GH therapy following a critical period of adipose and muscle development during infancy14). Therefore, it is important to evaluate the effects of early GH therapy on the physical findings and neurodevelopment of infants and toddlers with PWS.
In 1 study including similar-aged children with PWS who had received or not received long-term GH therapy, the strongest evidence to date showed that GH therapy, when started early in life, beneficially and significantly altered the natural history of PWS by reducing body fat and improving muscle strength physical function, and lipid profiles without adverse effects57). Thus, physicians and families can favorably weigh the sustained long-term value of GH treatment in infantile patients with PWS. Some experts advise that GH treatment should be started at a low dose such as 0.25-0.30 mg/(m2 · day) or 0.009-0.012 mg/(kg · day) and increased during the first weeks and months to reach the standard replacement GH dose of approximately 1.0 mg/(m2 · day) or 0.035 mg/(kg · day) with the monitoring of clinical effects, such as sleep apnea and increase in the level of IGF-I, particularly if there is a clinical suspicion of overtreatment (edema, worsening or new development of snoring, headache, and/or acromegalic clinical features)51). In recent years, the diagnosis of PWS during infancy has increased. At the Samsung Medical Center, there are nearly 180 patients with PWS, and 86 patients were diagnosed during infancy. Although children younger than 2 years of age are not covered by health insurance in Korea, GH therapy has been started in infants after obtaining informed consent from their parents in recent years. Their growth curve and development relative to chronologic age have improved with GH therapy. There have been no severe side effects. However, a long-term follow-up of the patients treated with GH may be needed.
3. Mechanism of the effects of GH therapy in infants with PWS
In a randomized, controlled trial, administration of GH to infants and toddlers with PWS normalized height, increased accrual of lean body mass, and reduced the percent body fat after 1 year of treatment. The 2-year data showed that the accumulation of excess body fat was delayed and reduced but not prevented, similar to an earlier study reporting a body fat SDS of +3 after 2.5 years of GH treatment60). The age at independent walking was younger than typical for the syndrome, 23.3±4.8 months in this study and 24.1 months in a previous study61). Subjectively, the GH-treated infants and toddlers were reported to be more alert and energetic by their families. An increased rate of language and cognitive development in the treated group was noted. It is unclear whether these results are solely from the known effects of GH therapy on muscle tone. However, the concomitant increase in head growth suggests that a central nervous system effect may also play a role14). In 1 study, IGF-I levels increased rapidly during GH treatment from below the normal range to the high-normal range62). IGF-I receptors have been localized in several areas of the human brain, indicating that IGF-I may have a neuroregulatory role in the central nervous system63). Theoretically, IGF-I may directly influence the central nervous system or GH might induce local IGF-I expression in brain tissue, thereby improving psychomotor development62). Another possible explanation for the improvement in mental development during GH treatment might be that because of the improved motor development, children are able to sit, stand, and walk independently, enabling them to explore and interact with the environment, resulting in subsequent improvement in mental development62).
In another study, after 1 year of GH treatment, there was a significant improvement in both mental and motor development in PWS infants and toddlers compared to randomized controls62). Head circumference increased from low-normal to normal during GH treatment. Children with an initially lower motor developmental age improved more than children with a higher initial motor developmental age, indicating that early treatment with GH might be beneficial. In the general population, there is evidence that intelligence tends to be higher in subjects with a greater head circumference, and brain growth during infancy and early childhood appears to be more important than that during fetal life in determining cognitive function64).
4. Concerns in GH therapy
Children with PWS are at risk of developing sleep-disordered breathing secondary to both deficient autonomic sleep control and upper airway obstruction. It has been suggested that GH exacerbates preexisting gas-exchange deficiencies in 3 ways: by stimulating adenotonsillar hypertrophy65), increasing the basal metabolic rate with a resultant rise in oxygen demand66), and normalizing previously decreased hydration with an augmentation of the volume load67). A causal relationship between GH and sudden death has not been demonstrated, although important concerns over safety have been raised. Concerns over possible worsening of obstruction are justified in the obese group, and patients with PWS require careful dietary, ENT, and respiratory evaluation before starting treatment with GH. Selected cases may benefit from noninvasive ventilatory support.
Complications and Prognosis
Patients with PWS can develop complications associated with hypogonadism (osteoporosis/pathological fractures) and obesity due to hyperphagia and hypometabolism (secondary to hypopituitarism), predisposing these patients to premature death from cardiorespiratory failure. They can also develop slipped capital femoral epiphyses/hip dysplasia, sleep apnea, cor pulmonale, and type 2 diabetes mellitus. Various neoplasias have been rarely reported in patients with PWS. In addition, because of binge-eating episodes, patients may have choking episodes (requiring the Heimlich maneuver), acute gastric dilation with risk of gastric necrosis, and food poisoning from the consumption of contaminated food69).
Patients with PWS frequently reach adulthood and are able to function in a group home setting, performing vocational work or attending community college classes. Diminished sensitivity to pain and diminished capacity to vomit may delay the diagnosis of underlying diseases (e.g., appendicitis). Complications from hypogonadism (e.g., osteoporosis/pathological fracture), behavioral issues (e.g., temper tantrums, stubbornness, and psychoses), and morbid obesity (e.g., type 2 diabetes mellitus and cor pulmonale) may shorten life expectancy and affect the quality of life. Patients with PWS can be mainstreamed into the classroom environment. They require additional speech therapy to enhance verbal skills and should have additional physical activity periods in place of rest periods. These individuals require a structured environment and may need a smaller classroom size for individual attention. Older children with PWS may enter vocational programs (with avoidance of food preparation). Some adults have attended community colleges.
Conclusion
The complex genetics, etiology, multiple phenotypes, and evolving natural history of PWS suggest that a multidisciplinary professional, parental, societal, and environmental approach to the management of these patients is required for overcoming the many challenges of reducing morbidity and mortality and improving patient quality of life. GH replacement therapy in PWS has resulted in drastic improvements in the phenotype, body composition, and self-image of treated individuals. However, much is yet to be learned about this complex disorder. It has been 27 years since the genetic region responsible for PWS was first identified, yet we still do not know the precise gene(s) responsible for the phenotype. Recent data suggest a key role of the HBII-85 snoRNA gene, and in the near future, researchers should uncover its gene targets, leading to improved treatments.
Acknowledgment
This work was supported by the Samsung Medical Center Clinical Research Development Program grant (CRS 105-45-2), a grant from the Korean Health 21 R&D project, Ministry of Health and Welfare (01-PJ10-PG6-01GN15-0001), the Samsung Biomedical Research Institute grant (SBRI C-A6-403-1, C-B0-206-1), and In-Sung Foundation for Medical Research.
Many thanks to YoungBae Sohn, Sung Won Park, Se-Hwa Kim, and Sung-Yoon Cho from the Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, for supporting this work.
Special thanks to Se-Chan Seo, a patient with mucopolysaccharidosis II, for drawing a valuable figure.