A case of pseudohypoaldosteronism type 1 with a mutation in the mineralocorticoid receptor gene
Article information
Abstract
Pseudohypoaldosteronism type 1 (PHA1) is a rare form of mineralocorticoid resistance characterized in newborns by salt wasting with dehydration, hyperkalemia and failure to thrive. This disease is heterogeneous in etiology and includes autosomal dominant PHA1 owing to mutations of the NR3C2 gene encoding the mineralocorticoid receptor, autosomal recessive PHA1 due to mutations of the epithelial sodium channel (ENaC) gene, and secondary PHA1 associated with urinary tract diseases. Amongst these diseases, autosomal dominant PHA1 shows has manifestations restricted to renal tubules including a mild salt loss during infancy and that shows a gradual improvement with advancing age. Here, we report a neonatal case of PHA1 with a NR3C2 gene mutation (a heterozygous c.2146_2147insG in exon 5), in which the patient showed failure to thrive, hyponatremia, hyperkalemia, and elevated plasma renin and aldosterone levels. This is the first case of pseudohypoaldosteronism type 1 confirmed by genetic analysis in Korea.
Introduction
Pseudohypoaldosteronism type 1 (PHA1) is a rare disease of mineralocorticoid resistance characterized by renal salt wasting, dehydration and failure to thrive in the neonatal period1, 2). The cardinal laboratory abnormalities of PHA1 are hyponatremia, hyperkalemia, and metabolic acidosis despite elevated plasma renin activity and aldosterone levels. There are two different genetic forms of PHA1, an autosomal dominant form (AD-PHA1) and an autosomal recessive form (AR-PHA1), as well as secondary PHA1, which is usually associated with urological problems2, 3). AD-PHA1 is caused by mutations of the NR3C2 gene, which encodes the mineralocorticoid receptor (MR). In addition, patients with AD-PHA1 manifest a renal tubule-restricted target organ defect that includes mild salt loss during infancy and a gradual improvement with advancing age4). Conversely, patients with the autosomal recessive form of PHA1 (AR-PHA1), which is caused by mutations of the amiloride-sensitive epithelial sodium channel (ENaC) gene, manifest severe salt wasting resulting from multiple mineralocorticoid target tissues including the sweat glands, salivary glands, respiratory tract epithelial cells and the kidneys2). Unlike AD-PHA1, AR-PHA1 has poor prognosis and requires life-long salt supplementation3).
In Korea, there have been several case reports of PHA15-12); however, none of these have been confirmed by genetic analysis. Here, we report a case of a 25 day-old boy with typical clinical features of PHA1 and a de novo heterozygous mutation in the NR3C2 gene (c.2146_2147insG in exon 5).
Case report
A 25 day-old male infant was admitted to our hospital due to poor weight gain and dehydration. The patient was born at full term with a birth weight of 2.63 kg (3-10th percentile). There were no perinatal problems, and the family history was unremarkable. Upon admission, his height was 49 cm (3-10th percentile), his weight was 2.46 kg (<3rd percentile), and his head circumference was 34.5 cm (10-50th percentile). Additionally, his blood pressure was 75/41 mmHg, pulse rate was 137/min, respiratory rate was 45/min, and body temperature was 36℃. The patient was alert at the time of admission, although his anterior fontanelle was slightly sunken and skin turgor was decreased. He had grossly normal external genitalia and other physical examination findings were unremarkable.
The initial laboratory examinations revealed the following: serum sodium, 122 mmol/L; serum potassium, 6.3 mmol/L; serum chloride, 91 mmol/L; blood urea nitrogen, 36 mg/dL; serum creatinine, 0.8 mg/dL; plasma renin activity, 114.1 ng/mL/hr (normal range 1-2.5); aldosterone, 9,840 pg/mL (normal range 50-194). The venous blood gas analysis revealed metabolic acidosis (pH 7.295, pCO2 41.3 mmHg, and HCO3- 20.1 mmol/L). The spot urine sodium, potassium, chloride and creatinine levels were 16 mmol/L, 9.7 mmol/L, 18 mmol/L and 3.3 mg/dL, respectively. Neonatal screening tests revealed a normal serum 17-hydroxyprogestenone level. Renal ultrasonography showed no abnormal findings. Genetic analysis revealed a heterozygous c.2146_2147insG (p.E716GfsX28) mutation in exon 5 of the NR3C2 gene that his parents did not have (Fig. 1).
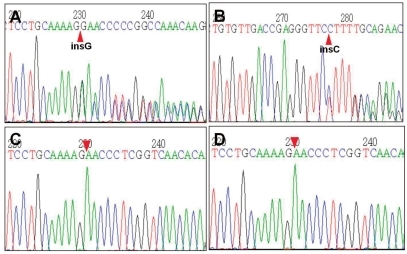
Partial sequencing data of the NR3C2 gene of the patient and his parents. The patient had a heterozygous c.2146_2147insG (p.E716GfsX28) mutation in exon 5 of the NR3C2 gene (panel A, 5'>3' sense sequences; panel B, 3'>5' complementary sequences). His parents (panel C, his mother; panel D, his father) did not have the insertion mutation.
The patient's serum electrolytes levels and renal function returned to normal with initial parenteral hydration and sodium supplementation for two days, and he then showed gradual weight gain with subsequent oral sodium chloride (Na+ 7-10 mEq/kg/day) supplementation. At the age of 6 months, oral sodium supplementation was discontinued at the outpatient clinic. The patient is currently nineteen-months-old and has resumed normal growth and development.
Discussion
Since PHA1 was first described in an infant with severe salt wasting syndrome by Cheek and Perry13) in 1958, there have been a number of case reports and recent molecular genetic studies that have identified two genetically different forms of PHA1, AD-PHA1 and AR-PHA13, 4, 14, 15).
Mineralocorticoid plays a crucial role in the regulation of fluid or electrolyte balance and blood pressure16). Aldosterone is secreted in response to hypovolemia or hyperkalemia and increases the activity of ENaC in the kidneys by binding to intracytoplasmic mineralocorticoid receptor (MR)2). Aldosterone then stimulates reabsorption of sodium, as well as excretion of potassium and hydrogen in the distal nephron. Disruption of the intracellular MR signaling cascade, by either MR or ENaC gene mutation, leads to salt wasting and causes PHA1, i.e., hyponatremia, hyperkalemia and metabolic acidosis1, 16).
Since ENaC is expressed not only in the distal nephron, but also in the colon, sweat glands, salivary glands and lung epithelial cells, AR-PHA1, which is caused by mutations in one of the three subunits of the ENaC2, 14), presents salt wasting from these various organs as well as the kidneys17). Moreover, the salt loss in AR-PHA1 is severe and does not improve with age; therefore, patients require massive sodium supplementation throughout life.
AD-PHA1 is caused by loss-of-function mutations of the NR3C2 gene1, 2, 18, 20), and a significant number of the reported mutations are de novo mutations as shown in the present study21-23). Although mutant receptors may exert some dominant negative effects on the wild-type receptors, haploinsufficiency is known to be sufficient to cause AD-PHA11, 2, 22). When compared to AR-PHA1, the salt loss in AD-PHA1 is less severe, restricted to the kidneys, and improves spontaneously with age. Although mild in its course, it has been reported that AD-PHA1 can be associated with a high infant mortality rate. Geller et al.21) reported a number of unexplained deaths in infants at risk for AD-PHA1, which suggests that AD-PHA1 is potentially fatal to neonates. Therefore, early diagnosis and prophylactic salt supplementation for neonates at risk for AD-PHA1 is essential2). Exogenous mineralocorticoid treatment has no effect in patients with AD-PHA1, and patients usually respond well to sufficient salt supplementation19). The spontaneous resolution of symptoms after infancy in patients with PHA1 can be explained by (1) the change in diet from low-sodium human breast milk to higher salt diet and (2) maturation of sodium reabsorption function of the renal tubules with increasing age2, 4). However, high aldosterone levels may persist even after normalization of other laboratory findings, including serum electrolyte levels in all cases1).
Our patient presented with typical clinical findings of PHA1 during the neonatal period and showed spontaneous improvement after 6 months of age. NR3C2 gene analysis revealed a de novo frame-shifting insertion mutation (heterozygous c.2146_2147insG in exon 5). There have been several case reports of PHA1 in Korea5-12); however, this case is the first one that has been confirmed genetically (Table 1). Molecular genetic studies can provide accurate diagnosis as well as basic data for genetic counseling and understanding of the pathogenesis of PHA1.
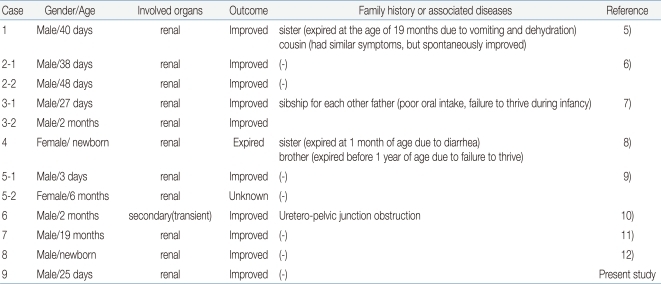
Case Reports of Pseudohypoaldosteronism Type 1 (PHA1) in Korea
Acknowledgment
This study was supported by a grant from the Korea Healthcare Technology R&D Project, Ministry for Health, Welfare and Family Affairs, Republic of Korea (A080588).