Recent advances in the diagnosis and management of childhood acute promyelocytic leukemia
Article information
Abstract
Since the successful introduction of all-trans-retinoic acid (ATRA) and its combination with anthracycline-containing chemotherapy, the prognosis for acute promyelocytic leukemia (APL) has markedly improved. With ATRA and anthracycline-based-chemotherapy, the complete remission rate is greater than 90%, and the long-term survival rate is 70-89%. Moreover, arsenic trioxide (ATO), which was introduced for APL treatment in 1994, resulted in excellent remission rates in relapsed patients with APL, and more recently, several clinical studies have been designed to explore its role in initial therapy either alone or in combination with ATRA. APL is a rare disease in children and is frequently associated with hyperleukocytosis, which is a marker for higher risk of relapse and an increased incidence of microgranular morphology. The frequency of occurrence of the promyelocytic leukemia/retinoic acid receptor-alpha (PML/RARα) isoforms bcr 2 and bcr 3 is higher in children than in adults. Although recent clinical studies have reported comparable long-term survival rates in patients with APL, therapy for APL in children is challenging because of the risk of early death and the potential long-term cardiac toxicity resulting from the need to use high doses of anthracyclines. Additional prospective, randomized, large clinical trials are needed to address several issues in pediatric APL and to possibly minimize or eliminate the need for chemotherapy by combining ATRA and ATO. In this review article, we discuss the molecular pathogenesis, diagnostic progress, and most recent therapeutic advances in the treatment of children with APL.
Introduction
Acute promyelocytic leukemia (APL) is a subtype distinct from all other acute myeloid leukemias (AMLs) with respect to the clinical, morphologic, cytogenetic, and molecular characteristics of a highly curable disease. These include the presence of life-threatening consumptive coagulopathy at diagnosis, high sensitivity of leukemic blasts to anthracyclines, and a balanced reciprocal translocation between chromosomes 15 and 17, t(15;17)(q22;q21), which results in a fusion between the promyelocytic leukemia (PML) gene on chromosome 15 and retinoic acid receptor-alpha (RAR alpha; RARα) on chromosome 17. The ability of leukemic promyeloblasts to undergo differentiation into neutrophils with all-trans retinoic acid (ATRA) depends on the presence of the PML-RARα fusion gene in leukemic cells1, 2).
ATRA-based chemotherapy as a type of targeted therapy contributes to the best relative overall prognosis among pediatric AML subtypes, with a remission rate greater than 90% and a 5-year disease-free survival of approximately 80%3-13). Moreover, arsenic trioxide (ATO), which was introduced in 1994 in APL, induces complete hematologic, cytogenetic, and molecular remissions via the dual effects of inducing partial differentiation and apoptosis of APL blasts14-21). Single ATO trials have demonstrated excellent remission rates in relapsed patients with APL14, 15), and more recently as front-line therapy either alone or in combination with ATRA16-18). In pediatric cases, when single agent ATO was used in newly diagnosed pediatric APL patients, remission rates of 86% and 3-year EFS of 75% were reported19-21). However, there is very limited experience with the combination of ATRA and ATO as initial therapy in children with newly diagnosed APL. Treatment strategies continue to evolve rapidly, with a focus on minimizing cytotoxic chemotherapy and introducing ATO as part of initial therapy. This review will discuss the incidence, molecular pathogenesis, diagnostic and therapeutic progress, and future therapeutic perspectives of APL in children.
Epidemiology
APL accounts for only 5-8% of pediatric AML cases. Approximately 600-800 children and adolescents develop acute leukemia each year in the United States22). In contrast to other subtypes of AML, which are equally represented across ethnic and racial groups, APL incidence varies widely among nations, with an increased incidence of APL in children from Italy and Central and South America23).
The age distribution of patients with APL differs from other forms of AML. While APL is uncommon in the first decade of life, the incidence increases during the second decade reaching a plateau during early adulthood; then the incidence remains constant until it decreases after 60 years of age. APL appears to be slightly more common in females.
Clinical presentations
APL more frequently presents with a higher incidence of hyperleukocytosis in children than in adults (approximately 40% in children vs. 20-25% in adults)24). It is caused by an increased incidence of microgranular (M3v) morphology, constituting up to 25% of pediatric cases, and a more frequent occurrence of the PML-RARα isoforms bcr 2 and bcr 3 in children25). Initial leukocyte count is the most important prognostic factor in patients with APL, and children with WBCs higher than 5,000/mm3 or 10,000/mm3 have a high risk of relapse26). Compared to other types of AML, in APL, leukopenia is significantly more common, hepatosplenomegaly is less common, and central nervous system (CNS) involvement of leukemia is rare.
Bleeding secondary to disseminated intravascular coagulation (DIC) is a unique presentation at diagnosis or after the initiation of cytotoxic chemotherapy in patients with APL. It can cause a 10-20% incidence of early death due to hemorrhage27). The risk of DIC is higher in patients with the microgranular variant of APL. Starting treatment with a differentiation agent (e.g., all-trans retinoic acid [ATRA]) plus supportive care as soon as the diagnosis is suspected, irrespective of definitive cytogenetic or molecular confirmation, is important to rapidly improve the coagulopathy. Although the mechanism for APL-induced DIC is not fully understood, bleeding coagulopathy is caused by the enhanced fibrinolytic activity involving 3 factors-tissue factor, cancer procoagulant, and increased annexin II receptor expression on the surface of the leukemic promyelocytes28). Tissue factor forms a complex with factor VII to activate factors X and IX. Cancer procoagulant activates factor X. Annexin II receptor binds plasminogen and its activator, tissue plasminogen activator, thus increasing plasmin formation.
Molecular pathogenesis
More than 95% of APL patients have a balanced reciprocal translocation between chromosome 15q22 and 17q21, which results in a fusion between the PML gene and RARα called PML-RARα. RARα is encoded on the long arm of chromosome 17 (Fig. 1A)29) and is a member of the retinoic acid (RA) nuclear receptor family that acts as a ligand-inducible transcription factor by binding to specific response elements (RARE) at the promoter region of target genes. It is mainly expressed in hematopoietic cells and has an important role in regulating gene expression. In the absence of RA, retinoic acid receptor (RAR) forms heterodimers with the retinoid X receptor (RXR), and the RA alpha gene is bound by the nuclear corepressor factor, thereby causing recruitment of the corepressor complex, chromatin condensation, and transcriptional repression. In the presence of RA, the genes are activated, and terminal differentiation of promyelocytes occurs. The promyelocytic gene (PML) is encoded on the long arm of chromosome 15 is thought to be involved in apoptosis and tumor suppression29).
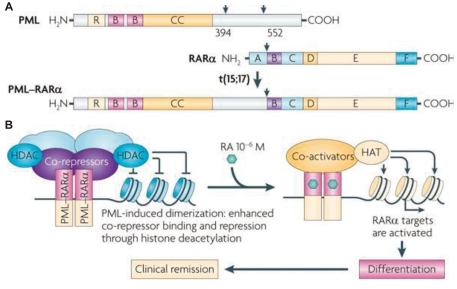
A) Structure of the promyelocytic leukemia (PML), retinoic acid receptor-α (RARα), and the PML-RARα fusion proteins. RING (R), B boxes (B), and coiled-coil (CC) domains in PML are indicated. The RARα DNA-binding domain (C) and hormone-binding domain (E) are shown. A, B, D, and F are other regulatory domains. PML-RARα retains the functional domains of both proteins, allowing dominant-negative activities on both PML and RARα. B) PML-RARα homodimers bind and repress RARα targets through enhanced recruitment of corepressors. Retinoic acid (RA) converts PML-RARα into an activator and restores differentiation, yielding clinical remission. HAT, histone acetyltransferase; HDAC, histone deacetylase (From de The et al, Nat Rev Cancer 2010;10:775-83).
The breakpoint in chromosome 17 is consistently found in intron 2. However, there are 3 possible isoforms caused by the translocation based on the PML breakpoint location in chromosome 15. The 3 breakpoints in PML occur at intron 3 (L form), intron 6 (S form), and exon 6 (V form). Compared with the L form, the S form is associated with a shorter remission duration and overall survival (OS)30).
PML-RARα homodimerizes via the PML coiled-coil domain and causes the RAR to bind more tightly to the nuclear corepressor factor and histone deacetylases (HDACs) on RARα target genes, thereby enforcing DNA methylation. Therefore, physiological doses of RA cannot activate the genes and induce repression of RA signaling, which blocks transcription and differentiation of granulocytes. Pharmacological doses of RA convert PML-RARα into a transcriptional activator, thus enhancing the expression of crucial RARα targets and restoring normal differentiation (Fig. 1B)29).
In about 5% of cases, alternative rearrangements of chromosome 17q21 with other gene partners are observed (Fig. 2). These include promyelocytic leukemia zinc finger (PLZF)/RARα t(11;17)(q23;q21), nucleophosmin (NPM)/RARα t(5;17)(q35;q12-21), nuclear mitotic apparatus (NuMa)/RARα t(11;17)(q13;q21), and/or signal transducer and activator of transcription 5b (STAT5b)/RARα t(17;17)(q11;q21). All of these rearrangements are ATRA-sensitive, except for PLZF/RARα, which is not sensitive to ATRA or ATO31).
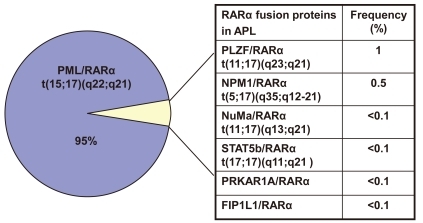
Acute promyelocytic leukemia-associated RARα fusion protein.
Diagnostic work-up
1. Morphology
APL cells are morphologically different from normal promyelocytes. These cells are larger and typically have creased, folded, bilobed, or kidney-shaped nuclei with heavy azurophilic granules and bundles of Auer rods (faggots). There are several main morphologic variants of APL. (a) The most common variant is the hypergranular or typical form, which accounts for approximately 75% of cases. The cytoplasm of these promyelocytes typically contains densely packed, bright-pink, reddish-blue, or dark-purple granules and frequently contains Auer rods, which are clumps of granular material containing lysosomes, peroxidase, lysosomal enzymes, and large crystalline inclusions. Bundles of Auer rods, called faggot cells, are also seen occasionally. (b) The microgranular (hypogranular or agranular; M3v) variant accounts for approximately 25% of cases32). These cells have bilobed nuclei and fine, dusky granules in the cytoplasm; Auer rods are rare. (c) The hyperbasophilic microgranular subtype has an increased nucleocytoplasmic ratio and strongly basophilic cytoplasm with cytoplasmic budding that mimics micromegakaryocytes. There are few granules and no Auer rods. (d) PLZF-RAR alpha (M3r) variants have regular, condensed chromatin in the nucleus. There are fewer granules, and Auer rods are rare compared with the hypergranular subtype.
2. Immunophenotype
Compared with other subtypes of AML, APL has distinctive immunophenotypic features such as frequent expression of CD13, CD33, and CD19; low or negative expression of CD34; and rare expression of CD117, HLA-DR, and CD11b. Unlike normal promyelocytes, APL cells express abnormally low levels of CD1533). Fifteen percent of APL cases express the CD56 antigen at diagnosis, and in some studies, this has been associated with worse outcome31).
The pattern of hypogranular variants of APL is more heterogeneous, with a higher percentage of cells expressing the T-cell antigen CD2, the stem cell marker CD34, HLA-DR, and CD56. CD19 has also been correlated with the M3v form. CD34 expression was suggested as a reliable marker to distinguish between M3v and classic APL. Hyperbasophilic APL expresses HLA-DR, CD33, CD13, HLA-DR, CD34, CD2, and CD9.
3. Molecular and genetic diagnosis of APL
Identification of the specific genetic lesion in APL cells with t(15;17)(q22;q12);PML-RARA is feasible at the chromosome, DNA, RNA, and protein level using conventional karyotyping, fluorescence in situ hybridization (FISH) for PML/RARA, Southern blot, reverse-transcriptase polymerase chain reaction (RT-PCR) for PML-RARA RNA, and anti-PML monoclonal antibodies. In general, cytogenetic, FISH, and molecular analyses are attempted simultaneously. Karyotyping is highly specific and is an essential part of standard work-up, but it has the following disadvantages: good-quality metaphases are required, it is time-consuming, and cryptic rearrangements leading to PML-RARA will be missed (false negatives). However, rare molecular subtypes of APL, such as t(11;17), t(5;17), and other additional coexistent cytogenetic abnormalities can be detected. FISH is less expensive, highly specific, faster than conventional cytogenetics, and can usually be completed within 24 h; however, it does not provide information about the PML-RARA isoform detected and therefore cannot be used for the molecular monitoring of residual disease. In contrast, RT-PCR provides information on the PML breakpoint location (bcr 1, bcr 2, and bcr 3) in addition to the advantages of FISH. This is the current "gold standard" for confirming the diagnosis of APL and is very useful for assessing response and follow-up monitoring of minimal residual disease (MRD). However, RT-PCR has a long turn-around time (approximately 2 days) and can have both false positives (contamination artifacts) and false negatives (due to poor RNA yield). RT-PCR should be performed at presentation to precisely characterize the amplification target, even in patients with APL confirmed by cytogenetics and/or FISH. Southern blot is a highly specific DNA-based method; however, it is now rarely used for APL diagnosis because it is laborious. Finally, indirect immunofluorescence or immunohistochemistry using anti-PML monoclonal antibodies is an easy method that allows for the rapid identification of the characteristic microgranular nuclear distribution of the PML protein in approximately 2 h, but is not widely used. However, sometimes the staining pattern of non-APL cases can be difficult to interpret.
Initial approach for suspected APL
APL usually presents as abrupt onset. The early mortality rate is high (10-20%) because of hemorrhage from a characteristic coagulopathy. When a diagnosis of APL is suspected by the characteristic morphology of leukemic population, the disease should be managed as a medical emergency, because hemorrhagic complications within the first hours and days are frequent and can be lethal. The treatment must be started immediately with attention of following instructions. First, start ATRA therapy as soon as possible after reviewing peripheral blood and bone marrow, and even before definitive cytogenetic or molecular confirmation of the diagnosis has been made. Second, the administration of supportive care with fresh frozen plasma and cryoprecipitate or platelet transfusions should be started to maintain fibrinogen and platelets above 100-150 mg/dL and 20,000-50,000/mm3, respectively, until all clinical and laboratory signs of coagulopathy have disappeared. Immediate treatment with ATRA on the same day as diagnosis is suspected rapidly improves coagulopathy. Third, confirmation of APL diagnosis should be performed at the genetic level by demonstrating the presence of t(15;17) and PML-RARα.
Induction therapy for newly diagnosed APL patients
1. Standard front-line therapy
After the introduction of ATRA in APL therapy, Huang et al34) were the first to report on the effectiveness of tretinoin for patients with APL in the late 1980s. In the early trials, tretinoin was administered as a single agent to induce remission followed by consolidation chemotherapy, but several studies showed that ATRA-induced remissions were short-lived and had no effects on survival35), suggesting the need to add chemotherapy during consolidation to obtain prolonged survival. More recent studies have shown that patients receiving ATRA followed by chemotherapy had significantly better outcomes than patients treated with chemotherapy alone. Furthermore, the European APL group showed improved outcomes in patients treated with simultaneous ATRA and chemotherapy than with sequential use36). Since then, there have been several other multicenter trials based on the same concept, and simultaneous administration of ATRA and anthracycline-based chemotherapy is currently considered the standard induction treatment for newly diagnosed patients. This combination therapy gives a better quality of complete remission (CR) with rates of 90-95% and is more effective in controlling ATRA-induced leukocytosis in newly diagnosed patients with APL.
The type of chemotherapy that is more advantageous for improving CR rates with ATRA is not clearly defined. Concerning the role of cytarabine, a randomized study was defined the patients treated with DNR+cytarabine had a better outcome than those treated with DNR alone39). Another group observed that cytarabine gave a benefit to patients with WBC counts greater than 10,000/mm3 and was associated with better outcome38). Therefore, this issue remains controversial. However, many clinicians still use ARA-C in combination with ATRA and anthracyclines. For anthracycline, most clinicians prefer IDA to DNR, but there is no prospective data showing any advantage for comparing IDA versus DNR. Possible therapeutic options for APL are summarized in Table 1.

Possible Options for treatment of Acute Promyelocytic Leukemia
Limited studies have reported therapeutic results using combinations of ATRA and anthracycline-based chemotherapy in children with APL3-13). In general, these studies showed improved outcome, with CR rates above 90% and a disease-free survival rate of 77%. These outcomes in children are comparable with those reported in adult patients. However, pseudotumor cerebri are observed more frequently in children than in adults during ATRA treatment, and a high cumulative dose of anthracycline resulted in increasing late cardiac toxicity in children. To decrease these risks, some groups have used a reduced dose of ATRA (e.g., 25 mg/m2 instead of 45 mg/m2), and a decreased incidence of pseudotumor cerebri with excellent results was reported at an ATRA dose of 25 mg/m2/d than with 45 mg/m2/d in children and adolescents with APL3-6). More prospective, randomized, large clinical trials are needed in children with APL.
Most newly diagnosed APL patients achieve CR with ATRA-based induction chemotherapy followed by a complete molecular remission after consolidation therapy. Resistance to primary therapy in APL is extremely rare or absent, and this situation causes suspicion of incorrect diagnosis39). The most frequent cause of induction failure is early death due to fatal cerebral hemorrhage. APL coagulopathy will clearly be improved by the addition of ATRA. The outcome of clinical trials in pediatric patients with APL treated with ATRA and anthracycline-based chemotherapy is summarized in Table 2.

Clinical Outcome of Newly Diagnosed Children with Acute Promyelocytic Leukemia by using ATRA-based Chemotherapy
2. Role of ATO in front-line therapy
ATO has proven to be another effective agent for APL therapy. The mechanism of action for ATO, which differs from that of ATRA, includes the induction of partial differentiation at low concentrations and apoptosis at higher concentrations in APL cells. Since ATO has been shown to induce remission in the treatment of relapsed APL, with observed remission rates of more than 80%14, 15), several trials have explored the role of this agent in front-line therapy16-21). Previous studies have shown that ATO with or without ATRA is equally effective in inducing remission in newly diagnosed patients with APL, and a CR rate of more than 85% and 5-year EFS and OS of more than 90% were reported16-21). Zhou et al21) reported an 89.5% CR rate, an 83.9% 5-year OS, and a 72.7% EFS in 19 children with newly diagnosed APL treated with single-agent ATO. These results demonstrate the high efficacy and minimal toxicity of ATRA+ATO treatment for newly diagnosed APL in long-term follow up, suggesting a new potential front-line therapy for de novo APL patients.
Despite these promising results, the current recommendation for induction therapy in newly diagnosed APL is the standard approach with ATRA+chemotherapy. It might appear reasonable to consider the possibility of a randomized study comparing conventional treatment using ATRA+chemotherapy with ATO alone or ATRA+ATO to minimize the chemotherapy in patients with good or intermediate-risk disease.
3. Evaluating response to induction therapy
An initial response to induction therapy can be determined with bone marrow aspiration and biopsy performed after the patient has recovered their absolute neutrophil count (>1,000/mm3) and platelet count (>100,000/mm3). However, morphologic, cytogenetic, and molecular follow up should be interpreted with caution in patients with APL during and soon after induction because the morphologic features of bone marrow receiving ATRA-containing regimens show a relatively hypercellular pattern. This reflects the residual differentiation of leukemic cells, and the persistence of atypical promyelocytes is occasionally detectable several weeks after the start of induction therapy with ATRA. Such features may be misleading as resistant disease. In these cases, ATRA-based treatment should be continued to obtain terminal differentiation of blasts and achieve hematological remission. To summarize, early morphologic, cytogenetic, and molecular assessments at the end of induction have no value for therapeutic decision making, and such assessments should wait until blasts have terminally differentiated and hematological remission has been achieved.
4. Consolidation therapy
The treatment strategy, including the type of consolidation and drug combination for post-remission therapy, is more controversial. Nevertheless, there is a consensus on the administration of at least 2-3 courses of anthracycline-based chemotherapy with or without Ara-C39). More than 90-99% of patients achieved molecular remission after receiving 2-3 cycles of anthracycline-based chemotherapy40). A small fraction of patients have persistent molecular disease at the end of consolidation and is needed to required additional, more intensive treatments, including hematopoietic stem cell transplantation (HSCT) when feasible.
As in induction, the role of cytarabine also remains controversial in consolidation. A randomized clinical trial reported comparable CR rates for ATRA+idarubicin (IDA) and ATRA+daunorubicin (DNR+cytarabine regardless of differences in risk group26). However, there is a trend in favor of cytarabine administration for high-risk of relapse patients (initial WBC count >10,000/mm3) on the basis of published results, which have demonstrated an improved antileukemic effect for the cytarabine-included regimens and a reduced relapse rate in high-risk patients40-42). In summary, the combination chemotherapeutic drugs including cytarabine is recommended for patients with an initial WBC count >10,000/mm3 who are at major risk of relapse.
Although the benefit provided by the addition of ATRA to chemotherapy for consolidation is not confirmed, the GIMEMA and PETHEMA groups showed a statistically significant reduction in relapse risk by adding ATRA at the standard dose (45 mg/m2/d for adults and 25 mg/m2/d for children) in conjunction with 3 courses of consolidation chemotherapy for 15 days24, 42).
It is not certain whether there is a benefit provided by the addition of ATO for post-remission therapy. Significantly better EFS and OS were reported with the use of ATO as post-remission treatment before the standard consolidation regimen with 2 more courses of ATRA +DNR or ATO43). This result provides further support for the use of ATO for purpose of eliminating of chemotherapy in consolidation.
5. Maintenance therapy
The potential use of maintenance treatment is an unresolved question in APL patients with molecular-negative disease at the end of consolidation. European APL group conducted the advantage of ATRA for maintenance therapy in APL and showed a lower relapse rate by combination of intermittent ATRA, methotrexate (MTX) and 6-mercaptopurine (6MP)44). But, more recent report by a Japanese study showed no any benefit of ATRA in maintenance therapy45) Studies to determine the role of ATRA-based maintenance therapy is still ongoing. The recommended schedule is to administer ATRA for 15 days at intervals of 3 months for 2 years. A therapeutic algorithm for newly diagnosed patients is proposed in Fig. 3.
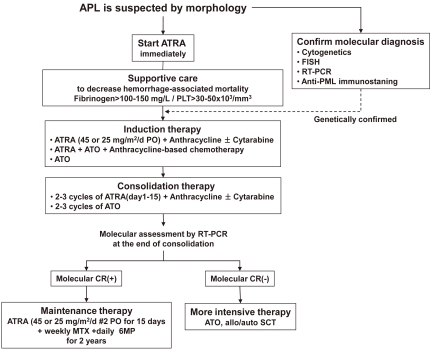
Therapeutic algorithm for newly diagnosed patients.
6. Response assessment and MRD monitoring
Unlike the assessment of response after induction therapy, molecular assessment of response after the completion of consolidation can be clinically relevant and is important to determine the risk of relapse, because the achievement of molecular remission at this time is a major treatment objective in APL patients. RT-PCR of PML-RARα in bone marrow with relatively low sensitivity, detecting 1 leukemic cell in 103 to 104 cells, is usually used to detect molecular remission. Patients who test PML-RARα positive after consolidation have poor prognosis and a potential risk of rapid progression to overt relapse. To rule out false positives, BM is repeated for MRD assessment within 2 weeks. More aggressive salvage therapy with ATO and/or chemotherapy and allogeneic HSCT should be considered for these patients. In contrast, patients who test negative for PML-RARα after consolidation have a low risk of relapse and should undergo maintenance therapy. We need to consider how often molecular monitoring after consolidation therapy is appropriate. It is reasonable to determine the frequency of monitoring for MRD according to the initial risk of relapse. Conversion to PCR positivity for PML-RARα during remission is highly predictive of subsequent hematologic relapse and underlines the prognostic value of MRD.
Treatment-associated adverse events
1. APL differentiation syndrome
A potentially life-threatening complication called differentiation syndrome (DS), formerly known as retinoic acid syndrome, occurs in 2.5-30% of newly diagnosed APL patients receiving ATRA treatment. This syndrome has also been described in patients with relapsed APL who received induction therapy with ATO. DS usually develops during induction therapy with ATRA and/or ATO, typically during the first 30 days of therapy. It is present neither during consolidation or maintenance therapy using 2 drugs, nor during ATRA treatment in non-APL malignancies. Leukemic APL cells might play a fundamental role in the development of DS. The clinical symptoms of DS are unexplained fever, cough, dyspnea, peripheral edema, weight gain, pleural fluid retention, interstitial pulmonary infiltrates, hypotension, pericardial effusion, and acute renal failure. This is often mistaken for fluid overload with pleural effusion or pneumonia, congestive heart failure, and diffuse alveolar hemorrhage. The syndrome usually develops within 2 weeks of therapy and is commonly associated with a rapidly rising leukocyte count. The probable risk factors for DS are a high leukocyte count and a rapidly increasing leukocyte count. This syndrome is associated with increased cytokine release from differentiating myeloid cells and modulation of adhesion molecules on the surface of aggregated APL blasts and vascular endothelial cells as a result of exposure to ATRA, which results in capillary leak and increased adherence to capillary endothelium. Steroid rapidly reduces these aggregations of APL cells. If DS is suspected or definitive, specific therapy with dexamethasone 0.3 mg/kg/dose twice daily in children (10 mg twice daily in adults) should be started immediately because this syndrome can be potentially life threatening, with a mortality rate as high as 9%46). Early introduction of chemotherapy has significantly reduced this incidence of DS. A temporary discontinuation of ATRA and/or ATO is indicated in severe case of DS. ATRA can be restarted in most cases once the syndrome has resolved.
2. Pseudotumor cerebri
Another complication of induction therapy with ATRA is pseudotumor cerebri (PTC), which is more commonly observed in children with APL than in adults, and occurs in 5-15% of those enrolled in clinical trials5, 6). It is characterized by increased intracranial pressure, severe headache, nausea, vomiting, and in severe cases by vision disturbances and papilledema. The mechanism of CNS toxicity by ATRA is not clear, but an excess of retinol is postulated to lead to a sustained increase in the choroidal secretion of cerebrospinal fluid or a decrease in its absorption by the arachnoid villi. Treatment of PTC is a temporary discontinuation or dose reduction of ATRA and the administration of dexamethasone, osmotic diuretics, and analgesics. To decrease these risks, some groups have used a reduced dose of ATRA (e.g., 25 mg/m2 instead of 45 mg/m2), and a decreased incidence of PTC with excellent results has been reported at an ATRA dose of 25 mg/m2/d than with 45 mg/m2/d in children and adolescents with APL4).
Prognostic factors in APL
The most important prognostic factor in patients with APL is leukocyte count at initial presentation. Children with WBCs higher than 5,000/mm3 or 10,000/mm3 have a high risk of relapse26). FMS-like tyrosine kinase 3 internal tandem duplications (FLT3/ITD), which are mutations in a tyrosine kinase receptor, have recently been considered as another prognostic indicator in pediatric APL. The incidence of FLT3/ITD mutations is 22.2% in non-APL patients, compared with 34.9% in children with APL47). These mutations are often associated with hyperleukocytosis at diagnosis and higher mortality during induction therapy. The microgranular variant form of APL is frequently associated with FLT3/ITD. However, the clinical impact of these mutations on relapse rate or OS is not yet entirely clear, and more clinical studies are needed.
Treatment of relapsed APL patients
Although more than 80% of pediatric patients with newly diagnosed APL can be cured with ATRA-based therapy and primary drug resistance is extremely uncommon, 10-15% of patients have a relapse, and 20-30% of those have high-risk APL. Therefore, it is difficult to conduct randomized studies with ATRA-based chemotherapy and/or ATO-based treatment as salvage therapy. Only limited data are available, including small series. Several treatment regimens have been utilized to treat relapsed disease, including ATRA, anthracyclines, high-dose cytarabine, autologous or allogeneic HSCT, ATO, and gemtuzumab ozogamicin. However, in this setting, most physicians would recommend the use of ATO. The efficacy of ATO for the treatment of both newly diagnosed and refractory or relapsed patients with APL has been proven in adults and children14, 15). These studies reported a second CR rate of 80-90% and OS at 1-3 years of 50-70%. ATO is administered intravenously at a recommended dose of 0.15 mg/kg/d until hematologic remission, or for a maximum of 60 days. ATO is generally well tolerated, and only mild toxic effects have been reported in the majority of patients. However, more severe complications may occur, including DS and prolongation of the QT interval to a treatable degree. ATO can lead to molecular CR after relapse and can play a critical role prior to transplantation.
Since the introduction of ATO, an additional course of ATO+ ATRA is recommended after achieving a second CR as consolidation therapy, and is followed by a molecular response after 2 cycles of ATO, to determine the continuation of therapy or risk of relapse. In a patient who is at higher risk, such as persistent PCR positivity, allogeneic HSCT should be done. Regarding the choice of autologous or allogeneic HSCT, both therapeutic options are feasible and have comparable outcomes39).
Conclusion
Over the last 20 years since the introduction of ATRA, there have been remarkable advances at the laboratory and clinical level in treatment of APL. APL is the most curable disease and is a paradigm for successful targeted treatment. However, the treatment of APL in children is challenging because of the risks of early death and potential long-term cardiac toxicity resulting from the need to use high doses of anthracyclines. Moreover, information on the long-term outcome from randomized studies in children treated with ATRA and anthracycline-based chemotherapy is still limited. Up-front use of ATO, either in induction or consolidation, allows children to avoid conventional cytotoxic chemotherapy in the near future. Prospective, randomized large clinical trials are needed to address several issues in APL and to possibly minimize and eliminate chemotherapy by combining ATRA and ATO.