Transient neonatal diabetes mellitus caused by a de novo ABCC8 gene mutation
Article information
Abstract
Transient neonatal diabetes mellitus (TNDM) is a rare form of diabetes mellitus that presents within the first 6 months of life with remission in infancy or early childhood. TNDM is mainly caused by anomalies in the imprinted region on chromosome 6q24; however, recently, mutations in the ABCC8 gene, which encodes sulfonylurea receptor 1 (SUR1), have also been implicated in TNDM. Herein, we present the case of a male child with TNDM whose mutational analysis revealed a heterozygous c.3547C>T substitution in the ABCC8 gene, leading to an Arg1183Trp mutation in the SUR1 protein. The parents were clinically unaffected and did not show a mutation in the ABCC8 gene. This is the first case of a de novo ABCC8 gene mutation in a Korean patient with TNDM. The patient was initially treated with insulin and successfully switched to sulfonylurea therapy at 14 months of age. Remission of diabetes had occurred at the age of 16 months. Currently, the patient is 21 months old and is euglycemic without any insulin or oral hypoglycemic agents. His growth and physical development are normal, and there are no delays in achieving neurological and developmental milestones.
Introduction
Transient neonatal diabetes mellitus (TNDM) is a rare form of diabetes diagnosed in the first 6 months of life. TNDM is differentiated from permanent neonatal diabetes mellitus (PNDM) by its remission in infancy or early childhood, with a possible relapse during adolescence1). More than half of TNDM cases are associated with chromosomal anomalies of an imprinted locus at 6q242). Some cases of TNDM are caused by mutations in the KCNJ11 gene, which encodes the inwardly rectifying potassium-channel subunit (Kir6.2), and the ABCC8 gene, which encodes the sulfonylurea receptor 1 (SUR1) of the adenosine triphosphate (ATP)-sensitive potassium (KATP) channel that is expressed at the surface of pancreatic beta cells. Closure of the KATP channel causes calcium ion influx and exocytosis of insulin from pancreatic beta cells3). Mutations in the ABCC8 gene cause diabetes by reducing the channel's ability to close in response to increased ATP and reduced adenosine diphosphate concentrations1). Here, we describe the first case of a de novo ABCC8 gene mutation in a Korean patient with TNDM. This mutation predicts a codon change from arginine to tryptophan at residue 1183 (Arg1183Trp), which is located in a functionally critical region of the KATP channel in pancreatic beta cells.
Case report
A newborn male baby was referred from a primary clinic due to tachypnea and chest retraction after birth. The patient was born at 35 weeks gestation by cesarean section delivery with a birth weight of 2,300 g (25th-50th percentile). He was diagnosed with respiratory distress syndrome (RDS) and the RDS symptoms improved after 3 days with ventilator care. Glucose monitoring revealed hyperglycemia with a glucose level of >300 mg/dL on the 7th day of life. There was no history of gestational diabetes. Physical examination did not reveal any dysmorphic features or instability. He was not receiving parenteral nutrition, and sepsis markers were negative. Hyperglycemia persisted without metabolic acidosis or ketonuria. The serum C-peptide was 0.36 ng/mL (normal range, 1.0-3.5 ng/mL), and HbA1C was <1% (normal range, 2.1-7.7%). Autoantibodies associated with type 1 diabetes were not present. He had no abnormal findings in diagnostic imaging and on function studies of other endocrine glands, such as the thyroid and adrenal glands. He was the second child of two siblings, and his parents and brother were clinically unaffected and had never experienced diabetic symptoms.
The patient was started on subcutaneous neutral protamine hagedorn (NPH) and regular insulin, on which he was discharged to home following resolution of hyperglycemia. He remained NPH insulin-treated with a gradually increasing insulin requirement over the first year of life. At 14 months of age, the patient was prescribed glyburide (2.5 mg once daily) and was successfully transferred from insulin to sulfonylurea treatment with good glycemic control. HbA1C level dropped from 7.5% (mean: 7.3% on insulin) at start of sulfonylurea treatment to 5.9% at 12weeks after transfer. Hypoglycemia that developed several times during insulin therapy was not observed after the transfer onto sulfonylurea treatment. He entered remission at 16 months of age and we stopped his treatment. The patient currently exhibits normal growth and physical development and has not experienced any delays in achieving neurodevelopmental milestones.
The patient was initially screened for abnormalities in chromosome 6q24. Since no 6q24 abnormality was identified, mutation screening was performed by sequence analysis of the entire coding region of the KCNJ11 and ABCC8 genes. Genomic DNA was extracted from the peripheral blood of the proband and family members (Fig. 1A) by using a DNA extraction kit (SolGent, Daejeon, Korea). All participants provided written informed consent, and the study was conducted in compliance with the Institutional Review Board of Konyang University Hospital.
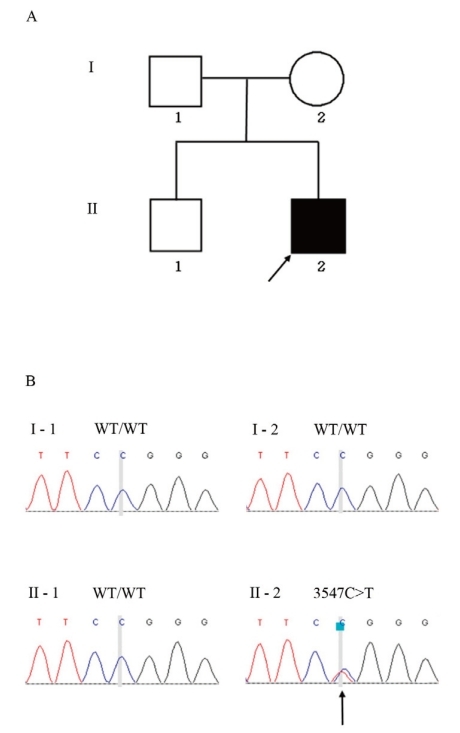
Family pedigree and mutation analysis. (A) Pedigree of the TNDM patient shows the proband (black symbol) indicated by an arrow and the unaffected family members (open symbols). (B) Identification of a de novo missense mutation in the ABCC8 gene in the TNDM patient. Electropherograms show the sequence encompassing the heterozygous transition mutation (c.3547C>T) in ABCC8 exon 28 in the patient (right lower panel) and the corresponding wild-type sequences in the normal family members.
Full exons of the KCNJ11 and ABCC8 genes were amplified to scan for mutations. Primers were designed from the introns surrounding the exons using Primer3 software (http://www.genome.wi.mit.edu/cgi-bin/primer). Primer sequences are available upon request. Polymerase chain reaction (PCR) was performed in a total volume of 25 µL containing 100 ng of genomic DNA, 10 pmol of each primer, and 2 × H-Taq Multiplex premix consisting of a PCR buffer, dNTP, MgCl2, and H-Taq DNA polymerase (SolGent) using a thermal cycler (GeneAmp PCR System 9700; Applied Biosystems, Foster City, CA, USA). The PCR conditions consisted of initial denaturation at 95℃ for 15 min, followed by 35 cycles at 95℃ for 20 s, 60℃ for 20 s, 72℃ for 30 s, and a final extension at 72℃ for 2 min. The PCR products were purified using a Millipore FB plate (Millipore, Billerica, MA, USA), and the nucleotide sequence was determined with an automatic genetic analyzer (ABI 3730XL) using the Big Dye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems, Foster City, CA, USA). The obtained sequences were then compared with the sequences from the control samples using SeqMan software (DNAStar, Madison, WI, USA), and the sequence variations were confirmed by analyzing both DNA strands.
Direct sequencing of ABCC8 exon 28 revealed a heterozygous C>T transition at nucleotide 3547 (Fig. 1B), leading to the substitution of a highly conserved, positively charged arginine with a neutral tryptophan at codon 1183 (Arg1183Trp). This mutation was absent in the patient's parents (Fig. 1B). Parental identity was verified by genotyping 15 short tandem repeat markers using a PowerPlex 16 system (Promega, Madison, WI, USA).
Discussion
Neonatal diabetes mellitus, an uncommon cause of hyperglycemia during the period just after birth, presents within the first 6 months of life (with an estimated incidence of 1 in 400,000-500,000 neonates) and may persist for 2 weeks or more. TNDM is a clinically defined subgroup affecting -50% of children with neonatal diabetes1, 2, 4). TNDM is differentiated from PNDM by its remission in infancy or early childhood. However, relapse in childhood or adolescence occurs in up to 50% of cases. Changes in insulin requirements and changes in the number of pancreatic beta cells have been implicated in the cause of the biphasic course, but the exact mechanism remains undetermined1). Recently, TNDM has been shown to result from activating mutations in SUR1 that reduce the ability of ATP to close the KATP channel5). Mutations in the ABCC8 gene are thought to account for about 10% of all cases of neonatal diabetes and frequently cause TNDM6).
In this study, we report the first case of a de novo mutation in the ABCC8 gene in a Korean patient with TNDM. This mutation is located in a region involved in linking the transmembrane domain to the nucleotide binding site in the SUR1 subunit of the pancreatic beta cell KATP channel7). Among the identified mutations of the ABCC8 gene, the Arg1183Trp mutant is very rare. Two previous studies described patients with the same mutation but different clinical phenotypes that included diabetic ketoacidosis and neurological features, such as seizures and severe developmental delay. These patients experienced a complete remission within 2-13 months1, 7). Our patient has not experienced any ketoacidosis or ketonuria. Patients with TNDM are less likely to develop ketoacidosis than patients with PNDM, and some studies showed absence of ketoacidosis in TNDM patients with 6q24 abnormalities2, 8). Lower incidence of diabetic ketoacidosis might be related to insulin resistance, which is associated with the onset of recurrence in the TNDM patients1, 9). At 21 months of age, our patient currently exhibits normal growth and physical development without seizures or delays in achieving neuro-developmental milestones, although he experienced a later remission than did previous cases.
Our patient had been receiving NPH insulin injections for glycemic control before the mutation analysis revealed his SUR1 mutation. Based on the analysis result, treatment with glyburide (a sulfonylurea) was initiated. Previous reports have indicated that oral sulfonylurea therapy in patients with mutations of the KATP channel genes, ABCC8 and KCNJ11, can result in glycemic control that is as good, or better, than that achieved with subcutaneous insulin injection6, 10-13). Our patient discontinued insulin (after 13 months of therapy) after a successful response to glyburide (2.5 mg per day). After entering remission at 16 months old, he is currently maintaining euglycemia without any insulin or oral hypoglycemic drugs.
In conclusion, we report the first de novo Arg1183Trp mutation in the ABCC8 gene in a Korean TNDM patient with a normal neurodevelopmental phenotype. Our findings have implications for the diagnosis, clinical management, and genetic counseling of a child who exhibits the phenotypic features of TNDM without a familial history of diabetes. We recommend that all patients with non-6q24 TNDM be tested for mutations in the ABCC8 and KCNJ11 genes in order to establish appropriate glycemic control by changing to oral sulfonylurea therapy.
Acknowledgement
The authors acknowledge Dr Deborah Mackay, Wessex Regional Genetics Laboratory, Salisbury District Hospital, Salisbury, UK, for testing chromosome 6q24 abnormalities.
This work was supported by the Korea Research Foundation Grant funded by the Korean Government (MOEHRD, Basic Research Promotion Fund) (KRF-2008-331-E00158).