Hypokalemic periodic paralysis; two different genes responsible for similar clinical manifestations
Article information
Abstract
Primary hypokalemic periodic paralysis (HOKPP) is an autosomal dominant disorder manifesting as recurrent periodic flaccid paralysis and concomitant hypokalemia. HOKPP is divided into type 1 and type 2 based on the causative gene. Although 2 different ion channels have been identified as the molecular genetic cause of HOKPP, the clinical manifestations between the 2 groups are similar. We report the cases of 2 patients with HOKPP who both presented with typical clinical manifestations, but with mutations in 2 different genes (CACNA1Sp.Arg528His and SCN4A p.Arg672His). Despite the similar clinical manifestations, there were differences in the response to acetazolamide treatment between certain genotypes of SCN4A mutations and CACNA1S mutations. We identified p.Arg672His in the SCN4A gene of patient 2 immediately after the first attack through a molecular genetic testing strategy. Molecular genetic diagnosis is important for genetic counseling and selecting preventive treatment.
Introduction
Hypokalemic periodic paralysis (HOKPP) is an autosomal dominant disorder characterized by recurrent episodes of reversible flaccid paralysis with concomitant hypokalemia and it is the most common form of primary periodic paralysis1). Symptoms begin in the first or second decades of life and the severity varies greatly in terms of frequency and duration of paralytic attacks. Well-known precipitating factors are a carbohydrate-rich meal, rest after strenuous exercise, emotional stress, and preceding infections2). The diagnosis of HOKPP is made if there are recurrent attacks of paralysis and hypokalemia, without any apparent cause for hypokalemia1). Patients with HOKPP can be grouped according to the underlying molecular genetic defects; HOKPP type 1 (online mendelian inheritance in man [OMIM] 170400) is due to mutations in the α-subunit of the DHP-receptor (CACNA1S)gene3) and HOKPP type 2 (OMIM 613345) is due to mutations in the α-subunit of the sodium channel (SCN4A) gene4). Although there are no significant differences in clinical presentation between the two groups, difference in response to treatment with certain SCN4A gene mutation has been reported5). We report two patients diagnosed with HOKPP type 1 and type 2 after identifying p.Arg528His mutation of the CACNA1S and p.Arg672His mutation of the SCN4A, respectively.
Case report
1. Case 1
An 11-year-old male presented to the emergency department with sudden onset quadriparesis. He had a history of transient lower extremity weakness fo llowing an upper respiratory infection 2 years ago. The patient was unable to move his limbs when he got up from a nap. The weakness was symmetric and involved both proximal muscles of upper and lower extremities. He denied strenuous exercise or a carbohydrate-rich meal. He had no dyspnea, dysphagia, or palpitation. He denied any recent weight change, diarrhea, chest pain, or heat intolerance. He had no significant past medical history, except the transient weakness 2 years ago. His family history revealed no similar episodes and no other significant illnesses other than the hypothyroidism of the patient's mother.
A clinical examination revealed normal vital signs (blood pressure, 120/66 mmHg; heart rate, 94 beats/min; respiration rate, 20 breaths/min; body temperature, 36℃), unremarkable cardiopulmonary examinations, and no thyroid enlargement. A neurological examination revealed symmetric upper and lower extremity weakness (grade III/V) and decreased deep tendon reflex. Sensory change, muscular atrophy, myotonia, and pyramidal tract signs were not present. Other neurological examinations including the cranial nerve examination were normal.
Laboratory tests including thyroid function test, routine chemistry (blood urea nitrogen [BUN], 11 mg/dL; creatinine, 0.8 mg/dL), electrolyte (sodium, 140 mmol/L; chloride, 106 mmol/L; bicarbonate, 20 mmol/L), complete blood count, urinalysis (pH 6.0), and urine chemistry were normal except for a serum potassium level of 1.9 (3.5 to 5.5 mmol/L). An electrocardiogram revealed typical findings of hypokalemia such as ST segment depression, T wave flattening, and U waves.
Intravenous potassium (20 mEq/10 mL) mixed with normal saline (240 mL, concentration 80 mEq/L) was infused over one hour while monitoring the electrocardiogram. After the intravenous replacement, oral potassium chloride supplementation was started (K-contin 600 mg BID). Serum potassium level after 4 hours of replacement was 4.0 mmol/L. The patient's symptoms completely resolved the next day with intravenous and oral potassium replacement. The electrocardiogram was also normalized. Studies to reveal the cause of hypokalemia at the outpatient clinic including serum renin and aldosterone were normal. After the first episode, the patient was started on an oral potassium supplement and had yearly attacks of paralysis with concomittant hypokalemia during the next 3 years. However, the number of attacks increased to twice per month in the recent few months without an identifiable cause. There was some improvement after adding spironolactone but with intermittent attacks. The paralytic attacks decreased after treatment with acetazolamide and the patients was attack-free for 3 months. Direct sequencing of exons 11 and 30 of CACNA1S and exon 12 of SCN4A were performed as an initial step of genetic diagnosis. A heterozygous c.1583G>A (p.Arg528His) mutation of CACNA1S exon 11 was identified (Fig. 1).
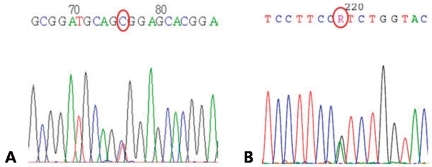
Polymerase chain reaction and direct sequencing of CACN1AS and SCN4A. (A) A heterozygous mutation in exon 11 of the CACNA1S gene causing p.Arg(CGC)528 His(CAC). (B) A heterozygous mutation in exon 12 of the SCN4A gene (skeletal muscle sodium channel α subunit) causing p.Arg(CGT)672His(CAT).
2. Case 2
A 15-year-old male presented to the emergency department with sudden onset lower extremity weakness. The patient awoke in the morning with weakness of both upper extremities. Lower extremities weakness appeared after he did strenuous exercise at school. Lower extremity myalgia was also present. The weakness was symmetric and it was more severe in the lower extremities. He denied a carbohydraterich meal. He had no dyspnea, dysphagia, or palpitation. He denied any recent weight change, diarrhea, chest pain, or heat intolerance. He had no significant past medical history, except an incidentally detected transient microscopic hematuria at the age of 7 and acute appendicitis at the age of 8. His family history revealed no similar episodes and no other significant illnesses.
A clinical examination revealed normal vital signs (blood pressure, 128/84 mmHg; heart rate, 70 beats/min; respiration rate, 20 breaths/min, body temperature, 36.6℃), unremarkable cardiopulmonary examinations, and no thyroid enlargement. A neurological examination revealed upper symmetric and lower extremity weakness (upper, grade III/V; lower, grade II/V) and an absent deep tendon reflex. Sensory change, muscular atrophy, myotonia, and pyramidal tract signs were not present. Other neurological examinations including the cranial nerve examination were normal.
Laboratory tests including thyroid function test, routine chemistry (BUN, 11 mg/dL; creatinine, 0.77 mg/dL), electrolyte (sodium, 140 mmol/L; chloride, 106 mmol/L; bicarbonate, 20 mmol/L), complete blood count, urinalysis (pH 6.0), and urine chemistry were normal except for a serum potassium level of 2.3 (3.5 to 5.5 mmol/L). An electrocardiogram revealed no abnormalities.
Intravenous potassium (20 mEq/10 mL) mixed with normal saline (490 mL, concentration 40 mEq/L) was infused over five hours while monitoring the electrocardiogram. The symptoms completely resolved a few hours after initiation of intravenous potassium replacement. After the intravenous replacement, oral potassium chloride supplementation was started (K-contin 600 mg BID). Serum potassium level immediately after the intravenous replacement and the next day was 3.0 and 5.2 mmol/L, respectively. Studies to reveal the cause of hypokalemia at the outpatient clinic, including serum renin and aldosterone, were normal. The patient was started on spironolactone and he had no further attacks for the last 4 months. Direct sequencing of exons 11 and 30 of CACNA1S and exon 12 of SCN4A were performed as an initial step of genetic diagnosis. A heterozygous c.2015G>A (p.Arg672His) mutation of SCN4A exon 12 was identified (Fig. 1).
Discussion
Typical clinical presentation of HOKPP is recurrent attacks of flaccid paralysis and hypokalemia beginning in the first two decades1). Two different genes, CACNA1S and SCN4A, are identified as responsible for the molecular genetic cause of HOKPP type 1 and type 2, respectively3,4). There are reports of genotype-phenotype correlations in certain mutations regarding the age of onset, frequency or duration of attacks, and triggering factors6). However, there were no differences between the CACNA1S and SCN4A group in terms of clinical presentations including the age of onset, attacks of flaccid paralysis and hypokalemia, precipitating factors, and outcomes2,7). The two patients in this report, who carried the mutation in different causative genes, also manifested a similar clinical presentation of HOKPP.
The exact pathophysiologic mechanisms how these mutations in CACNA1S and SCN4A genes produce prolonged muscle paralysis are not identified. However, paradoxical sustained muscle membrane depolarization in response to hypokalemia was demonstrated in both calcium channel and sodium channel mutations8-10). The voltagegated calcium channel located in the T-tubule of the skeletal muscle fiber (Cav 1.1) has a function of excitation and contraction coupling. The p.Arg528His mutation substitutes the positively charged arginine to lesser charged histidine, in the S4 segment of domain II, which is thought to be a voltage sensor of the channel. This substitution resulted in reduced calcium current density and rate of activation, finally causing reduced excitability11). Tricarico et al.12) showed reduced open probability and conductance states and hypothesized that abnormal potassium channel conductance resulted from altered calcium homeostasis. The "loss-of-function" feature of smaller currents and slower activation was demonstrated from cardiac rabbit mutant channel which contained mutation of segment 4/domain II13). The voltagedependent sodium channel of the skeletal muscle (Nav 1.4) has a function of action potential generation in response to membrane depolarization, which finally resulted in muscle contraction. Rudel et al.14) showed reduced excitability and increased sodium conductance in response to hypokalemia, and suggested these findings were the basic defects in HOKPP type 2. The p.Arg672His mutation resulted in reduced current and enhanced inactivation which finally caused impaired muscle contraction9,15). The authors of these reports suggested that this depolarization causes paralysis in both types of HOKPP8-12).
Although the clinical presentation did not vary significantly between the two groups of HOKPP, there are reports of different responses to treatment with acetazolamide in patients carrying a certain mutation of the SCN4A. The frequency and severity of attacks increased in some patients with p.Arg672Gly and p.Arg672Ser mutations of the SCN4A gene after treatment with acetazolamide4,5,16). On the contrary, patients with CACNA1S mutations, acetazolamide treatment was frequently effective2). In this report, patient 1 who harbored a CACNA1S mutation (p.Arg528His) showed good response to the potassium supplement having only 2 attacks in 3 years. However, the attacks occurred more frequently without any identifiable cause resulting in 2 to 3 attacks a month. Treatment with acetazolamide was effective in this patient. This finding is concordant with previous reports that acetazolamide is frequently effective in patients with the CACNA1S gene mutation. The treatment of HOKPP comprises management of acute paralysis and prevention of recurrent attacks. Acute attacks are treated with potassium replacement to normalize serum potassium and to shorten the duration of the attacks. An oral potassium supplement, acetazolamide, and spironolactone are used to prevent recurrent paralytic episodes with varying degrees of effectiveness2). Treatment with acetazolamide should be initiated carefully because of the risk of worsening symptoms in patients with certain genotypes.
Molecular genetic analysis of case series studies revealed that 65 to 80% of the HOKPP cases are caused by the mutation in the CACNA1S and SCN4A (CACNA1S, 55 to 70% and SCN4A, 8 to 10%)1,6,17). The mutations identified in this study are the most common mutation of the CACNA1S and the SCN4A gene1). A study done in Korea revealed that the CACNA1S and the SCN4A mutation were responsible for HOKPP in 75% and 25% of the patients, respectively, and the most frequent mutation was p.Arg528His for HOKPP17). These results are similar to those of Western case series reports6). Molecular genetic diagnosis is usually made after the clinical diagnosis of HOKPP in patients with periodic paralysis. Sternberg et al.2) proposed a testing strategy of sequencing exons 11 and 30 of CACNA1S and exons 12 and 18 of SCN4A, based on the frequency of pathogenic mutations identified. Patient 2 was diagnosed HOKPP 2 immediately after his first attack of paralysis and hypokalemia. Molecular genetic diagnosis must be made for all patients with HOKPP because genetic counseling is very important. Moreover, we can increase the diagnostic yield by applying the testing strategy based on the genotype frequency of the population.
The patients in this report presented with similar clinical manifestation of acute paralysis and hypokalemia. However, molecular genetic analysis revealed mutations in different causative genes (CACNA1S and SCN4A). Preventive treatment with acetazolamide was effective in patient 1 who harbored p.Arg528His mutation in the CACNA1S. Molecular genetic diagnosis was made after the first episode in patient 2 and p.Arg672His mutation was identified in the SCN4A.
Acknowledgement
This study was supported by a grant (A080588) from the Korea Healthcare technology R&D Project, Ministry for Health, Welfare and Family Affairs, Republic of Korea. The authors thank Mrs. MiSun Park for her English correction of the manuscript.