A case of partial trisomy 3p syndrome with rare clinical manifestations
Article information
Abstract
Partial trisomy 3p results from either unbalanced translocation or de novo duplication. Common clinical features consist of dysmorphic facial features, congenital heart defects, psychomotor and mental retardation, abnormal muscle tone, and hypoplastic genitalia. In this paper, we report a case of partial trisomy 3p with rare clinical manifestations. A full-term, female newborn was transferred to our clinic. She had cleft lip-plate, dysgenesis of the corpus callosum, patent ductus arteriosus, pulmonary hypertension, and severe right-sided hydronephrosis, associated with ureteropelvic junction obstruction. Cytogenetic investigation revealed partial trisomy 3p; 46,XX,der(4)t(3;4) (p21.1;p16). The karyotype of her father showed a balanced translocation, t(3;4)(p21.1;p16). Therefore, the size of duplication can be an important factor.
Introduction
Partial trisomy 3p is a rare chromosomal abnormality and it can be either de novo or inherited from one parent with a balanced translocation1). The common clinical features include early postnatal death, congenital heart defect, psychomotor retardation, hypoplastic genitalia and dysmorphic face, such as frontal bossing, temporal indentation, and hypertelorism1,2). The clinical findings associated with trisomy 3p are usually similar but not necessarily the same in all diagnosed cases1-8). The uncommon findings including holoprosencephaly, retinal change, and congenital anomalies of the kidney had rarely been reported6). We present a case of trisomy 3p with unique clinical characteristics, such as dysgenesis of the corpus callosum and significant ureteropelvic junction obstruction.
Case report
A newborn infant with dyspnea, cleft lip and palate, was transferred from the obstetric clinic just after birth. She was born via vaginal delivery, after 40+2 weeks of gestation, without any perinatal problems. Her mother was pluripara, with a history of 4 abortions (the reason was not known). The mother had not taken prenatal examination regularly. Both parents were normal in appearance and had no family history of any specific diseases.
The newborn suffered from asymmetric intrauterine growth retardation: her body parameters were the weight of 2,330 g (<10th percentile), the height of 43.5 cm (<10th percentile), and the occipitofrontal circumference of 33 cm (25 to 50th percentile). She showed dysmorphic facial features, such as temporal indentation, frontal bossing, hypertelorism, cleft lip and palate, large low set ears, and short neck (Fig. 1). The respiratory rate was 68 breaths/min, blood pressure was 62/31 mmHg and the oxygen saturation was 88% when supplying oxygen 3 L/min via the hood. The right-sided flank and abdominal area were distended. She had single transverse palmar creases in both hands. The external genitalia showed normal appearance. The neurologic examination revealed negative Moro, grasp and pupil reflex. Hypochromic gray irides, and pin point pupils were also observed. After endotracheal intubation (Mechanical ventilation mode: synchronized intermittent mandatory ventilation, FiO2 0.3; respiratory rate, 25/min; peak pressure, 15 cmH2O; positive end expiratory pressure, 4 cmH2O), the initial arterial blood gas analysis showed pH 7.19, pCO2 55.7 mmHg, pO2 88 mmHg, Sat 94%, bicarbonate 21.3 mEq/L. The chest radiography was normal. Echocardiography taken on the 1st day of age revealed patent foramen ovale (size, 5 to 7.3 mm), large patent ductus arteriosus (size, 3.4 mm), with bidirectional shunt (mainly left to right), grade II/IV tricuspid regurgitation (peak velocity, 3 m/sec), trivial mitral regurgitation and pulmonary hypertension. The brain ultrasonography performed on the 2nd day of age suggested a dysgenesis of the corpus callosum, with a cystic lesion on the right lateral ventricle. The brain magnetic resonance imaging exam conducted on the 16th day of age confirmed the dysgenesis of the corpus callosum and the left subependymal cyst (Fig. 2). Spine ultrasonography performed on the 30th day of age was normal. After respiratory supportive care, including inhalation of nitric oxide, extubation was conducted on the 9th day of age, and she breathed well by herself. Large secundum atrial septal defect, with large left-right shunt, and mild tricuspid regurgitation were observed on the follow-up echocardiography. There was no mitral regurgitation or patent ductus arteriosus. The follow-up abdominal ultrasonography showed marked dilatation of the right pelvocalyceal system, with parenchymal thinning (Fig. 3). Voiding cystourogram performed on the 17th day of age was normal. Tc99m-mercaptoacetyl triglycerine (MAG3) scan, with furosemide, was performed to check the function of the affected kidney, revealing right hydronephrosis, with significant ureteropelvic junction obstruction. The brain stem auditory evoked potential was absent to bilateral ear stimulation, suggesting bilateral acoustic nerve lesion. The newborn's metabolic screening, using tandem mass spectrometry, was normal. All markers for congenital infection were negative. At the 20th day of age, fever and pyuria developed. Klebsiella pneumoniae and Enterococcus faecium were identified in urine and blood cultures, respectively. The patient was treated with intravenous antibiotics (i.e., vancomycin and carbapenem for 10 days) until the negative result of the bacterial culture and clinical improvement was observed. The ophthalmologic examination revealed large disc and cup with retinal discoloration, a feature of cataract, but the exact diagnosis was not reached due to limitation of the examination methods. The light reflexes in both pupils remained sluggish until leaving hospital. The neurologic examination at the 25th day of age was relatively within normal range, except for the asymmetric Moro reflex, negative asymmetric tonic neck reflex, negative grasp reflex, and negative plantar reflex. She was fed by gavage, until she was discharged from our hospital at the 35th day of age.
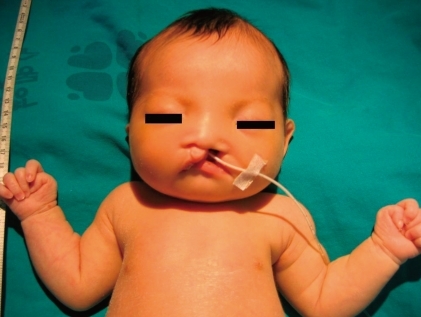
Temporal indentation, a square shaped face, telecanthus, hypertelorism, cleft lip and palate, downturned mouth corners, and short neck, are shown.
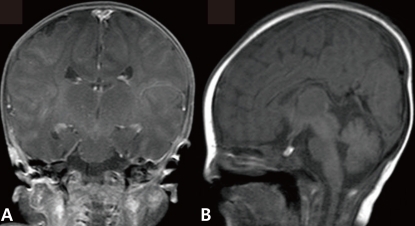
T1-weighted coronal (A) and sagittal (B) magnetic resonance images showed dysgenesis of the corpus callosum.

An abdominal ultrasonography revealed marked dilatation of the right renal pelvocalyceal system with parenchymal thinning, suggesting ureteropelvic junction obstruction.
The cytogenetic investigation of the patient using G-banding demonstrated a partial trisomy 3p (Fig. 4A). She had an extra genetic material on part of the chromosome 3 (unbalanced translocation). The break points were on the short (p) arm of the derivative chromosomes 3 and 4. The chromosomes from the mother were normal, but the father had a balanced translocation; t(3;4)(p21.1;p16) (Table 1, Fig. 4B). The patient didn't revisit our outpatient clinic because she had to live in her hometown.
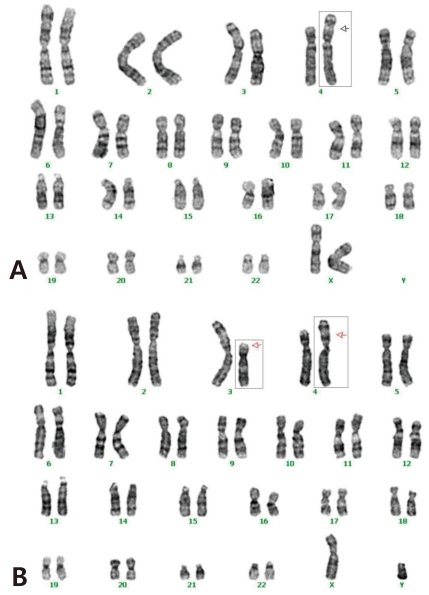
(A) Idiogram (Giemsa [GTG]-banding) of the patient. Karyotyping showed partial duplication of chromosome 3; 46XX,der(4)t(3;4)(p21.1;p16) pat. (B) Idiogram (GTG-banding) of the patient's father. Karyotype: 46XY,t(3;4)(p21.1;p16).

Results of Peripheral Blood Chromosome Analysis
Discussion
We present a case of partial trisomy 3p with unique clinical manifestations, including dysgenesis of the corpus callosum and significant ureteropelvic junction obstruction. These findings of our case have been rarely reported, although some authors had reviewed partial trisomy 3p cases and the pattern of anomalies according to the frequency of the features1,2,4).
The parental origin of major cases is usually maternal. Conte et al.1) noted that the majority of trisomy 3p cases are maternally derived. In our patient, the karyotyping of chromosomes showed partial duplication of the chromosome 3p and unbalanced translocation: 46,XX,der(4)t(3;4)(p21.1;p16)pat. Whether the carrier of the abnormal chromosomes is the father or the mother, however doesn't seem to be decisive in determining the clinical features1,2).
The derivative chromosomes, the size of duplication and the rearranged parts involved in translocation are varying in previous reports. Schinzel et al.3) reported a patient with translocation, t(3:4) (p23;q35). However, the clinical manifestations were not similar in our patient and in Schinzel's patient. The factors influencing the clinical features seem to be complex.
The carriers of balanced translocations are usually not symptomatic and are likely to conceive children with abnormalities8). Patients with unbalanced translocation usually show dysmorphic features and/or congenital anomalies9,10). It is essential to evaluate the karyotype of the parent whose child has multiple congenital anomalies. In addition, a long period of observation is required for detecting mental retardation.