X-linked recessive myotubular myopathy with MTM1 mutations
Article information
Abstract
X-linked recessive myotubular myopathy (XLMTM) is a severe congenital muscle disorder caused by mutations in the MTM1 gene and characterized by severe hypotonia and generalized muscle weakness in affected males. It is generally a fatal disorder during the neonatal period and early infancy. The diagnosis is based on typical histopathological findings on muscle biopsy, combined with suggestive clinical features. We experienced a case of a newborn who required intubation and ventilator care because of profound hypotonia and respiratory difficulty. The preliminary diagnosis at the time of request for retrieval was hypoxic ischemic encephalopathy, but the infant was clinically reevaluated for generalized weakness and muscle atrophy. Muscle biopsies showed variability in fiber size and centrally located nuclei in nearly all the fibers. We detected an MTM1 gene mutation of c.1261-1C>A in the intron 10 region, and diagnosed the neonate with myotubular myopathy. The same mutation was detected in his mother.
Introduction
Myotubular myopathy (MTM) is an X-linked congenital disorder (XLMTM) that is caused by MTM1 mutation1). In this condition, the small muscle fibers present a central area devoid of myofibrils and mitochondrial aggregates around the centrally located, often large nuclei2). Compared with other congenital myopathies, patients with XLMTM have a rather homogeneous clinical presentation3). The onset of clinical signs is typically at or near birth, and affected infants are areflexic and floppy, exhibiting respiratory difficulty that usually requires ventilatory support4). Respiratory insufficiency often complicates the clinical course and causes death among patients5). Most of these patients die during infancy or early childhood3).
The majority of mutations related to XLMTM have been identified in exons 3, 4, 8, 9, 11, and 126,7). Several cases have been reported since MTM was first described in 1986 in Korea8,9). However, only one case was reported with regard to the genetic analysis of MTM in the Korean population10). Here we report a case of myotubular myopathy in a neonate with a splice site mutation of MTM1 gene at the intron 10 region.
Case report
A 2,930 gm male was born at 37 weeks and 5 days gestation age via emergency cesarean section in a local hospital. He was the first child of healthy parents. Polyhydramnios and reduced fetal movements were noted during the antenatal period. During the therapeutic amniocentesis in response to polyhydramnios, placental abruption occurred and fetal distress was observed. His Apgar scores were 1-5-5 at 1, 5, and 10 minutes; he required resuscitation and intubation due to poor respiration.
Subsequently, he was transferred to Pusan National University Yangsan Hospital and was placed on ventilator support. His birth weight (2,930 g, 50th percentile), length (51 cm, 75th to 90th percentile), and head circumference (35 cm, 90th percentile) were within the normal range. He had no abnormalities based on the chest radiograph, but findings showed fractures on both his humerus which were splinted for treatment within one month (Fig. 1).

On infantogram fractures on both humeri were observed.
The child's initial features were similar with the effects of birth asphyxia. His birth condition presented severe hypotonia, sucking impairment, and the absence of spontaneous movements. He then required mechanical ventilation from birth throughout his confinement at the hospital due to apnea.
However, magnetic resonance imaging of the brain and electroencephalography showed no evidence of hypoxic ischemic encephalopathy. Postnatal blood gas analysis showed normal levels; echocardiography and screening blood tests for metabolic disease were also normal.
His generalized hypotonia with absent deep tendon reflexes continued. No neonatal reflexes could be elicited and his posture was extremely floppy-frog like characterized by a long face (Fig. 2). Tests for congenital hypotonic disorders, including spinal muscular atrophy, myotonic dystrophy, Pompe disease, Prader-Willi syndrome, and chromosomal disease were carried out. However, all tests yielded negative findings. Muscle biopsy of the left biceps was performed and myotubular myopathy was confirmed based on the findings characterized by large centrally placed nuclei in myofibers (Fig. 3). Peripheral blood samples for the gene study were obtained from the patient and from his mother for X-linked myotubular myopathy. A mutation c.1261-1C>A was identified in the intron 10 of the MTM1 gene in this case. His mother was also noted to have the same mutation (Fig. 4).
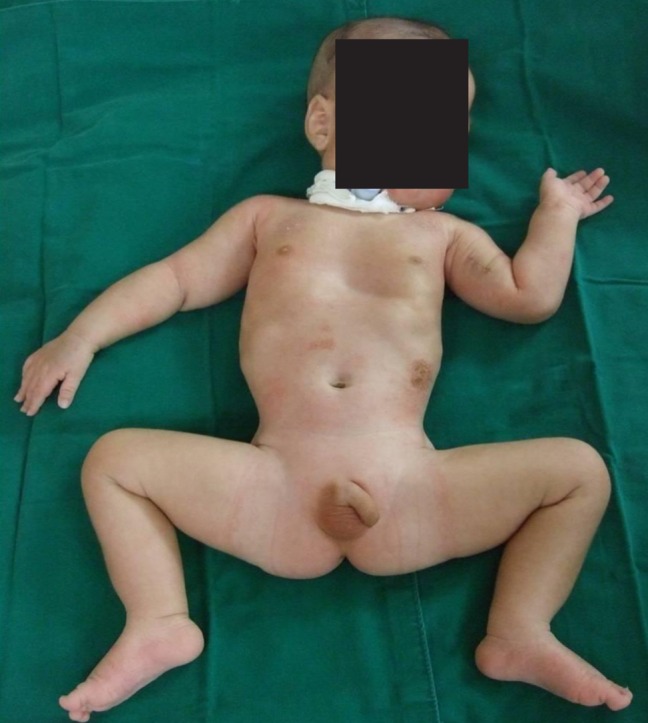
The patient manifesting a frog-like posture, long face, and undescended testes.
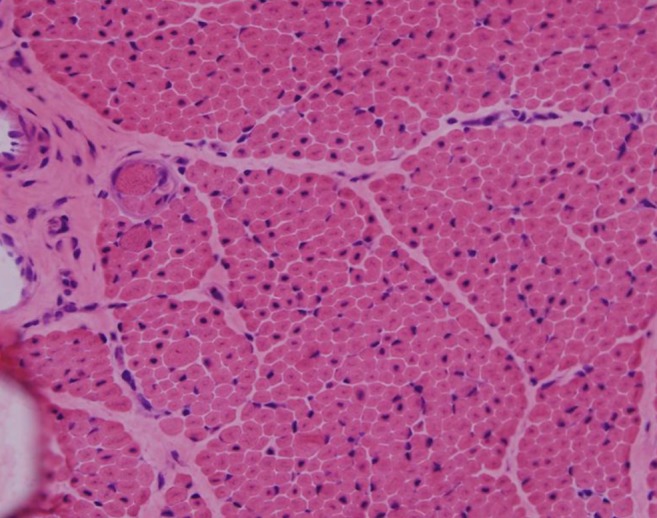
H&E staining (×200) under light microscopy showing centrally located nuclei in nearly all the fibers, with marked variation in fiber size.
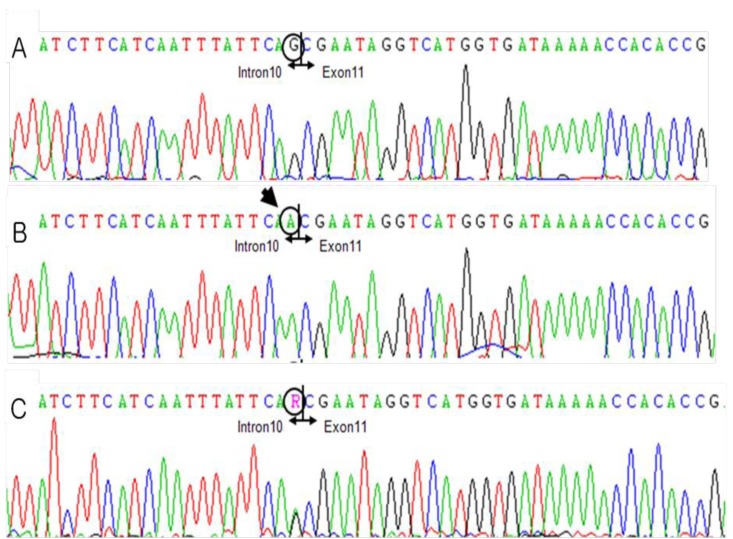
DNA sequence electropherograms of the patient and his mother, showing the identified MTM1 mutations in intron 10. (A) Normal DNA sequence, (B) patient's DNA sequence showing the c.1261-1G>A mutation, and (C) his mother's DNA sequence showing a heterozygous mutation of c.1261-1G>A.
He needed tube feeding for nutritional support and a tracheostomy for ventilator support. He was discharged after six months and was brought home with a mechanical ventilation support. He later died when he was seven months old due to respiratory failure.
1. DNA isolation and PCR reaction
Genomic DNA was isolated from peripheral blood using QuickGene DNA kit (Fujifilm life Science, Tokyo, Japan). To analyze of MTM1 gene mutation, polymerase chain reaction (PCR) was performed by use of fourteen sets of primers designed in intronic flanking region containing all exons referred to GenBank accession number NT_011726.13. PCR was carried out 30 cycles. After amplification, PCR mixtures were run on 1.2% agarose gel in the presence of ethidium bromide to verify the size and purity of PCR products.
2. DNA sequence analysis
After verifying that single specific PCR product was amplified, DNA sequencing was performed using the same primers used in PCR, and BigDye Terminatore V3.1 Cycle Sequencing kit (Applied Biosystems, Foster city, CA, USA) according to manufacturer's instructions. Reaction was performed 30 cycles. Electrophoresis and analysis of the reaction mixtures were done with ABI 3130xl Genetic analyzer.
Discussion
Mutations in the MTM1 gene encoding myotubularin cause XLMTM, a well-defined subtype of human centronuclear myopathy4). XLMTM is characterized by the early onset and presence of uniformly small muscle fibers with centrally placed nuclei3,4). Although centrally located nuclei can be found in many myopathies, clinical, genetic, and pathological factors can help distinguish these myopathies from XLMTM4).
Clinical characterization of XLMTM has been comprehensive and the disorder has been found to give rise to a severe phenotype in males. The most notable clinical manifestations of XLMTM are muscle weakness and associated disabilities. It is present at birth with marked weakness and hypotonia, external ophthalmoplegia and respiratory failure due to the weakness of the muscles responsible for respiration11). Signs of antenatal onset include reduced fetal movements, polyhydramnios and thinning of ribs as seen on chest radiographs11); However, these manifestations are rarely observed in other congenital myopathies. Birth asphyxia may be the presenting feature12). Many people tend to confuse XLMTM with birth asphyxia during initial detection.
The diagnosis of XLMTM has traditionally relied upon the identification of characteristic histopathologic changes in muscle samples from affected males. Because of the time required for molecular analysis of MTM1, the clinical diagnosis of XLMTM continues to rely upon muscle biopsy13). Hematoxylin and eosin-stained cross sections of XLMTM muscle show increased variability in fiber size, but this is generally not as extreme as what is seen in the dystrophic processes. The myofibers typically appear small and have a rounded profile. The cardinal histopathologic feature of XLMTM is the presence of hypotrophic myofibers with relatively large, centrally placed nuclei3). This case was also confirmed as XLMTM by muscle biopsy.
The X-linked nature of this disorder facilitated genetic analysis, with linkage studies assigning the gene to Xq28 in 199011). Laporte et al.1) isolated the MTM1 gene by positional cloning and named the gene product, 'myotubularin'. In 1997, de Gouyon et al.14) and Laporte et al.15) carried out a complete gene sequencing of 15 coding exons and flanking intron. In several series, mutations were detected in 60% to 90% of individuals with XLMTM14,15). The majority of mutations were identified in exons 3, 4, 8, 9, 11, and 126,7).
The first report concerning an XLMTM family with two affected infants diagnosed by muscle biopsy and gene analysis was released in Korea8). A nonsense mutation Arg486STOP was identified in exon 7 of the MTM1 gene in that family. Moreover, we detected another splice site mutation of MTM1 gene c.1261-1C>A at the intron 10 region in a present neonate with XLMTM.
Although, the diagnosis is confirmed by muscle biopsy, MTM1 mutation analysis is critical in consulting with the mother about prenatal family planning and antenatal genetic screening. Prenatal testing should still be offered to all women who might give birth to affected male infants3).
There is no known treatment for such disorder16). The management is essentially supportive and/or rehabilitative, requiring a multidisciplinary approach11,17). In the majority of cases, the course is fatal within the initial months of life, but a proportion of affected males infants may survive into their teens or beyond 11,18,19).
Floppy infants can be misdiagnosed with hypoxic ischemic encephalopathy at birth. Therefore, physicians who care for the newborn should consider the possibility that a neuromuscular disorder might be present in a hypotonic infant. When a floppy infant is regarded to have hypoxic ischemic encephalopathy based on a perinatal circumstantial evidence, an accurate diagnosis should be established by carrying out appropriate clinical procedures. If a neuromuscular disorder is suspected, a muscle biopsy should be done. A gene study of a patient diagnosed with MTM is essential for genetic counseling.
We report that a neonate initially misdiagnosed with hypoxic ischemic encephalopathy was diagnosed with a XLMTM with a splice site mutation of MTM1 gene at the intron 10 region.
Notes
No potential conflict of interest relevant to this article was reported.