Chronic intermittent form of isovaleric aciduria in a 2-year-old boy
Article information
Abstract
Isovaleric aciduria (IVA) is caused by an autosomal recessive deficiency of isovaleryl-CoA dehydrogenase (IVD). IVA presents either in the neonatal period as an acute episode of fulminant metabolic acidosis, which may lead to coma or death, or later as a "chronic intermittent form" that is associated with developmental delays, with or without recurrent acidotic episodes during periods of stress, such as infections. Here, we report the case of a 2-year old boy with IVA who presented with the chronic intermittent form. He was admitted to Asan Medical Center Children's Hospital with recurrent vomiting. Metabolic acidosis, hyperammonemia, elevated serum lactate and isovalerylcarnitine levels, and markedly increased urine isovalerylglycine concentration were noted. Sequence analysis of the IVD gene in the patient revealed the novel compound mutations-a missense mutation, c.986T>C (p.Met329Thr) and a frameshift mutation, c.1083del (p.Ile361fs*11). Following stabilization during the acute phase, the patient has remained in a stable condition on a low-leucine diet.
Introduction
Isovaleric aciduria (IVA) is characterized as abnormal leucine metabolism resulting from a deficiency in isovaleryl-CoA dehydrogenase (IVD), which is inherited in an autosomal recessive manner. The clinical presentation of IVA is highly variable, ranging from severely affected to asymptomatic. It can present either in the neonatal period as an acute episode of fulminant metabolic acidosis, which can lead to coma or death, or it can manifest later as the "chronic intermittent form," which is associated with developmental delays, with or without recurrent acidotic episodes during periods of stress, and infections1-4). The inability to detect this disorder early may lead to severe neurocognitive dysfunction, psychomotor retardation, or death3,4). Here, we report a child whose diagnosis was delayed until 2 years of age. IVA was confirmed by genetic testing, which revealed the two novel mutations.
Case report
The patient was the second baby of nonconsanguineous Korean parents. He was born after 38 weeks of gestation. His body weight was 3.86 kg (90th percentile) and his height was 50 cm (75th percentile). On the second day after birth, he developed an emetic episode and was admitted for paralytic ileus for 2 weeks. Newborn screening using tandem mass spectrometry on the fifth day of life demonstrated normal results. His growth profile was within the normal range, but speech and motor developmental delays were noted. His older sister was normal in intelligence. At the age of 1 year and 9 months, he was admitted to the emergency department due to recurrent vomiting. He was lethargic, and his urination had decreased. Weight loss was not noted. Tachycardia and respiratory difficulties were noted. His weight was 12 kg (50th percentile) and his height was 84 cm (50th percentile). His abdomen was soft and flat, but bowel sounds had decreased. He smelled of no particular odor.
Arterial blood gas analysis indicated metabolic acidosis with incomplete respiratory compensation: pH 7.27, CO2 19.5 mmHg, bicarbonate 8.8 mEq/L. The anion gap was considered high at 18 mEq/L (normal range, 7 to 16 mEq/L), as was his serum lactate level at 4.51 mmol/L (normal range, 0.56 to 1.39 mmol/L) and blood ammonia at 54 µmol/L (normal range, 11 to 32 µmol/L). Abdominal radiography demonstrated paralytic ileus. Abdominal ultrasound sonography showed no signs of bowel wall dilatation or intussusception. Notably, tandem mass spectrometry indicated an elevated isovaleryl carnitine level of up to 14.1 µmol/L (<1.4 µmol/L). He was transferred to Asan Medical Center Children's Hospital at the age of 1 year and 10 months. A Korean Child Developmental Inventory assessment at age 22 months documented his gross motor development at 16 months of age, fine motor skills at 16 months of age, personal social skills at 20 months of age, language skills at 11 months of age, and cognitive-adaptive skills at 22 months of age. A significantly elevated serum isovaleryl carnitine level was measured after overnight fasting. His urine organic acid profile determined using gas chromatography-mass spectrometry indicated a markedly increased concentration of isovalerylglycine (761.1 mmol/mol creatinine [Cr], normal range not indicated), but a normal concentration of 3-hydroxyisovaleric acid (6.5 mmol/mol Cr; normal range, 0.7 to 14.4 mmol/mol Cr).
Genetic testing for the IVD gene was performed using genomic DNA that was isolated from his peripheral leukocytes. The patient had compound heterozygous variants: c.[986T>C];[1083del] (p.[Met329Thr];[Ile361fs*11]) (Fig. 1). The p.Ile361fs*11 variant was inherited from his mother, whereas p.Met329Thr was inherited from his father. Both variants were previously unreported. The p.Ile361fs*11 variant is a frameshift mutation, generating a premature truncated protein. Two in silico programs, Polyphen (http://sift.bii.a-star.edu.sg/) and Sorting Intolerant From Tolerant (http://blocks.fhcrc.org/sift/SIFT.html), predicted p.Met329Thr as a pathogenic variant. Following stabilization during the acute phase, the patient is now doing well on a low-leucine diet and has not had a metabolic crisis in 4 months.
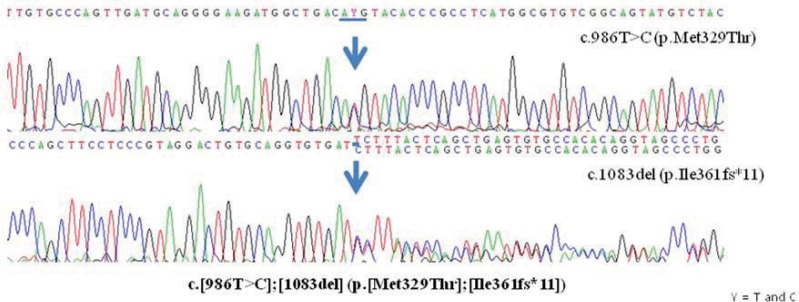
Sequence analysis of the isovaleryl-CoA dehydrogenase gene in the patient revealing a missense mutation, c.986T>C (p.Met329Thr), and a frameshift mutation, c.1083del (p.Ile361fs*11). The missense mutation was inherited from the patient's father, whereas the frameshift mutation was inherited from his mother. C, codon; T, thymine; C, cytosine; Met, methionine; Thr, threonine; Ile, isoleucine; del, deletion; fs*, frameshift.
Discussion
IVA is an autosomal recessive inborn error of branched-chain amino acids that is caused by a deficiency in IVD3-5). Patients with the acute neonatal type manifest with poor feeding, vomiting, seizures, and metabolic acidosis within the first 2 weeks of life. Patients with the chronic intermittent form develop intermittent metabolic derangement during episodes of stress, such as infections and developmental delays. Early diagnosis with a leucine-restricted diet is needed to prevent neonatal mortality and improve the neurologic and cognitive outcomes.
Our patient presented with the chronic intermittent form of IVA. He had already been experiencing acute exacerbations of IVA since the neonatal period. However, neonatal screening on the fifth day of life failed to detect isovaleryl carnitine, which is increased in IVA. This could have been due to decreased oral intake for the management of paralytic ileus. Severe acidosis, however, was diagnosed in this patient at 1 year and 9 months of age despite mild to moderate dehydration and a few episodes of vomiting, which often leads to metabolic alkalosis due to gastric acid loss and volume depletion. The presence of hyperammonemia, an elevated anion gap, metabolic acidosis, and developmental delays indicate that he might have an inborn error of metabolism that was exacerbated by gastroenteritis. Extensive analyses, including tandem mass spectrometry, plasma amino acid and urine organic acid profiles, and determination of elevated metabolites, isovaleryl carnitine, and isovalerylglycine were documented. With the implementation of newborn screening by tandem mass spectrometry, metabolic disorders can now be detected very early in life, which allows for the presymptomatic initiation of treatment in affected individuals and the prevention of mental retardation4). However, as in our case, IVA can be missed when newborns are on insufficient feeding.
The diagnosis of IVA is based on the results of enzymatic and genetic studies. Enzymatic assays for IVD that measure electron transfer flavoprotein reduction, tritium release, 14CO2 release, and 3-methylcrotonyl-CoA production by peripheral lymphocytes have been reported6-8). Although enzymatic assays were not performed on our current patient, IVA was confirmed by genetic testing, which can be directly performed when an enzyme assay is unavailable. To date, more than 40 heterogeneous mutations in the IVD gene have been reported in patients with IVA, including point mutations, frameshift mutations, and slice-site mutations (http://www.hgmd.org/)9). Missense and splicing mutations are the most common, but a small number of frameshift mutations have been reported. The correlation between phenotype and genotype is unknown. Recently, however, a common missense mutation, c.932C>T (p.Ala282Val), has been reported to be related to a mild phenotype of IVA10). In our present case, 2 novel mutations were identified. A frameshift mutation in exon 11 (p.Ile361fs*11), which was inherited from his mother and was predicted to produce a premature truncated protein. The other mutation is a missense mutation, p.Met329Thr. Although in vitro functional characterizations were not performed, this variant is believed to be a mutation because it is located on the trans-allele relative to the other mutation, which was inherited from his father, and is expected by in silico analysis to significantly affect the function of the protein.
Despite improvements in our understanding of the biochemistry and genetics of IVA, its management remains difficult. There are 2 goals for treatment. The first is the prevention of metabolic decompensation by careful clinical monitoring. Prevention of catabolism is the main therapeutic approach. Thus, during episodes of stress, IVA patients should receive adequate caloric intake while decreasing their leucine intake. The second goal is the long-term reduction in the production of isovaleryl-CoA via leucine catabolism. This can be accomplished using a low-protein or leucine-restricted and high-energy diet. Acute episodes of metabolic decompensation can present with emesis, lethargy, and signs of overwhelming acidosis. Under these circumstances, management should be based on exogenous protein restriction, the inhibition of endogenous catabolism by an adequate parenteral energy supply, high-dose carnitine and L-glycine, and vitamin supplementation4).
A few Korean cases of IVA have been reported11-13). Cheon and Lee12) reported the case of siblings with the chronic intermittent form of IVA. These 2 IVA patients were 3 and 8 years old, respectively, and had similar histories, including recurrent episodes of vomiting and lethargy that were resolved with supportive therapy. All of the previously reported cases were diagnosed by organic acid analysis. An IVA case with a neonatal form has not been reported previously. On the other hand, this is the second Korean case report confirmed by genetic testing. The first Korean case-series study by Lee at al.13) and our report indicate that the Korean patients with IVA exhibit the different mutation spectrum of the IVD gene from those in other populations, because most mutations in the two reports were novel and no common mutation was found. The incidence of IVA is not fully known, but several studies have estimated it at 1 in 67,000-365,00014,15). However, there may be additional persons with IVA who are presymptomatic or demonstrate a less severe form than those who have been reported in Korea. More effort is needed to increase the detection of IVA in Korea.
In conclusion, IVA is an inborn error of metabolism that can present as a severe neonatal form or as a late or intermittent form. Because neonatal screening may not be able to detect the mild form of IVA, extensive metabolic investigations should be considered for patients who exhibit the manifestations of the chronic intermittent form.
Acknowledgments
We thank the patient and his family for participating in this study, which was supported by a grant from the Korean Ministry for Health, Welfare and Family Affairs (No. A120367).
Notes
No potential conflict of interest relevant to this article was reported.