Necrotizing enterocolitis in newborns: update in pathophysiology and newly emerging therapeutic strategies
Article information
Abstract
While the survival of extremely premature infants with respiratory distress syndrome has increased due to advanced respiratory care in recent years, necrotizing enterocolitis (NEC) remains the leading cause of neonatal mortality and morbidity. NEC is more prevalent in lower gestational age and lower birth weight groups. It is characterized by various degrees of mucosal or transmural necrosis of the intestine. Its exact pathogenesis remains unclear, but prematurity, enteral feeding, bacterial products, and intestinal ischemia have all been shown to cause activation of the inflammatory cascade, which is known as the final common pathway of intestinal injury. Awareness of the risk factors for NEC; practices to reduce the risk, including early trophic feeding with breast milk and following the established feeding guidelines; and administration of probiotics have been shown to reduce the incidence of NEC. Despite advancements in the knowledge and understanding of the pathophysiology of NEC, there is currently no universal prevention measure for this serious and often fatal disease. Therefore, new potential techniques to detect early biomarkers or factors specific to intestinal inflammation, as well as further strategies to prevent the activation of the inflammatory cascade, which is important for disease progression, should be investigated.
Introduction
Necrotizing enterocolitis (NEC) was first described by Mizrahi et al.1) in 1965, and was classified into three stages based on the severity of the clinical presentation and treatment strategies by Bell et al.2) in 1978. Later, Walsh and Kliegman3) proposed the modified Bell's criteria, which subdivided each stage into A and B according to the clinical and radiologic signs and treatment strategies. There are inverse correlations between gestational age or birth weight and the incidence of NEC4,5), with, as demonstrated in multicenter and large population-based studies, its prevalence being 7%-11% in very low birth weight infants who weigh less than 1,500 g6,7).
Several experimental therapies have been tested in animal models and human trials4). Despite this progress, there has been no method to identify infants who would be ideal candidates for therapy or those who will progress to more severe stages of disease. This paper summarizes the recent advances in pathophysiology, the newly developed diagnostic approaches, and the current emerging therapeutic strategies for NEC.
Pathogenesis
Various combinations of risk factors, which may be based on genetic predisposition, have been demonstrated to increase the risk of mucosal and epithelial injury4).
1. Prematurity with impaired host defense
The most consistent and important risk factor is immaturity itself4,5,6,7). Immature motility, digestion, absorption, and circulatory regulation predispose the preterm infant to an increased risk of intestinal injury8). Especially, in these patients, the gastrointestinal (GI) host defense (physical barrier) and immunologic host defense, including intraepithelial lymphocytes and secretory IgA, are markedly impaired; and many biochemical factors such as lactoferrin, epidermal growth factor (EGF), heparin binding (HB)-EGF, transforming growth factor, insulin-like growth factor, oligosaccharides, and polyunsaturated fatty acids (PUFA), are deficient or absent4,8).
2. Enteral feeding
Enteral alimentation, especially formula feeding, is the second most important risk factor for NEC, with over 90% of NEC cases reportedly occurring after initiation of enteral feeding4). Since early trophic feeding is introduced in the care of extremely low birth weight infants, the onset is generally delayed for several weeks. The exact relationship between enteral feeding and NEC remains unclear, but the volume and rate of feeding advancement (overdistention may compromise splanchnic circulation), osmolarity, and substrate fermentation are considered important factors9,10). Short chain fatty acids, derived from fermentation of undigested carbohydrates by intestinal bacterial fermentation, have important effects on epithelial functions and maturation. In one animal study, overproduction/accumulation of short chain fatty acids in the proximal colon and/or distal ileum was shown to play a potential role in the pathogenesis of NEC11); however, no studies testing the effects of these molecules are available at the moment. Moreover, some studies have shown that deficiency of lactase or other brush border enzymes or accumulation of bile acid may also lead to mucosal injury4,12).
3. Bacterial colonization
In utero, the intestine is sterile, and no cases of NEC have been documented. The fact that NEC typically develops before extensive colonization of the GI tract by anaerobic organisms, and that probiotic therapy may prevent the disease, support the theory that microbes also play a key role in the initiation of intestinal injury4). Colonization of the intestines of a healthy breast-fed infant with anaerobic species such as Bifidobacterium and Lactobacillus occurs within 1 week of age13). However, a hospitalized, very low birth weight infant has less species diversity and fewer or absent anaerobes. This imbalance promotes pathologic proliferation and binding and invasion of otherwise nonpathogenic intestinal bacteria, and reduces the anti-inflammatory effects and mucosal defense from probiotic organisms14).
The lack of normal maternal-infant physical interaction during feeding interferes with normal intestinal colonization, which protects against the pathologic microflora in breast-fed premature infants4). Furthermore, contamination or colonization of the feeding tubes has been demonstrated to contribute to the development of NEC15). The most commonly isolated bacteria are Clostridium sp., Escherichia coli, Klebsiella sp., Staphylococcus epidermidis, and Enterobacter4). In particular, the use of infant formula contaminated with Enterobacter sakazakii has been associated with outbreaks of NEC16).
4. Intestinal ischemia/asphyxia
Intestinal ischemia due to hypoxia-ischemia was reported as an important risk factor for NEC in early studies, especially in term and late preterm infants, as the histologic hallmark of end-stage NEC is an "infarct" characterized by full thickness coagulation (ischemic) necrosis with scanty acute inflammatory cells (neutrophils) and a predominant lymphocytic infiltration17). However, intestinal ischemia is now unlikely to be considered the major perinatal event that contributes to the development of NEC. Instead, it is believed that it disturbs the balance in the microvascular tone related to the production of vascular regulators such as nitric oxide and endothelin, which likely play downstream roles in the pathogenic cascade of NEC18). Further, patent ductus arteriosus, another risk factor for NEC, results in intestinal hypoperfusion by diastolic steal phenomenon; and, in patients treated with indomethacin (a nonselective inhibitor of cyclooxygenase-1 and -2), focal intestinal necrosis or NEC may occur19).
5. Other factors
Although an association between the elective transfusion of packed red blood cells and NEC has been reported, the mechanism through which transfusion might be related to alteration in the intestinal blood flow or hypoxia-ischemia remains unclear20). H2 receptor antagonists, which are inhibitors of gastric acid, increase the gastric pH, which may enhance pathogenic bacterial growth and increase the risk of NEC21). As a genetic predisposition, the loss of the HB-EGF-like growth factor gene (which is essential for preservation of gut barrier function) was proposed in an animal study22). Furthermore, early use of postnatal steroids may cause intestinal perforation, independent of enteral feeding23); the use of umbilical artery or venous catheters does not seem to increase the risk of NEC24).
Pathophysiology
Recent advances in the understanding of NEC suggest that the epithelial barrier and innate immunity, together with the inflammatory response, play important roles in very preterm infants4). Intestinal mucosal stress (resulting from enteral feeding, bacterial products, or intestinal ischemia) and an inadequate host defense and repair (due to prematurity) can result in activation of the proinflammatory cascade, which is the final common pathway of intestinal injury and NEC. This cascade involves a balance between pro- and anti-inflammatory endogenous mediators, receptors, signaling pathways, and second messengers, and produces a variety of downstream effects that ultimately result in end-organ damage (Table 1,Fig. 1).
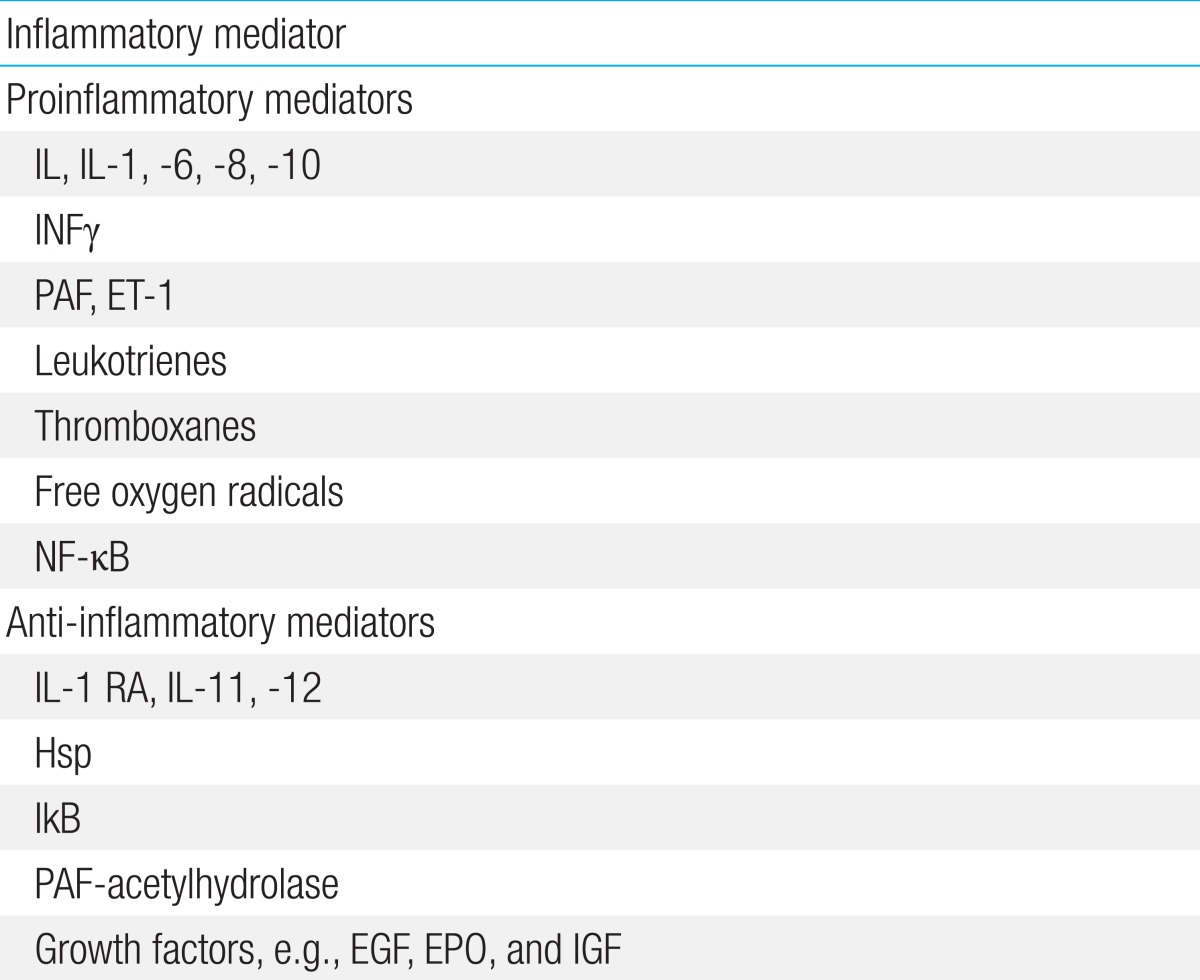
Inflammatory mediators involved in pathophysiology of necrotizing enterocolitis

Role of Toll-like receptor 4 (TLR4) in epithelial injury and repair mechanisms. Many factors related to prematurity such as infections, inappropriate enteral nutrition, antibiotics use, microcirculatory dysfunction, and hypoxia can induce epithelial injury. Hyperactivation of TLR4 enhances translocation of pathological bacteria across the epithelial barrier. Modified from Terrin et al. Biomed Res Int 2014;2014:54376527).
1. Epithelial barrier
In neonates, the intestinal mucosa presents a persistent equilibrium state between injury and repair. Injury to the intestinal mucosa may depend on a variety of conditions characteristic of prematurity, including hypoxia, infection, and starvation. Furthermore, microcirculatory dysfunction contributes to epithelial damage. In physiological conditions, healing of the epithelium begins immediately after an injury with mature enterocyte migration to the wounded area from healthy adjacent areas25). Subsequently, the proliferation of new enterocytes within the crypts of Lieberkühn completes the process of repair26). It has been recently suggested that NEC is associated with a marked inhibition in both enterocyte migration and proliferation, making the host uniquely susceptible to further injury, and that the loss of epithelial barrier allows translocation of pathogens from the intestinal lumen to the mucosa27).
2. Innate immunity: the role of Toll-like receptors
Innate immunity has been demonstrated to regulate the epithelial barrier in both experimental models and human cases of NEC. The specific Toll-like receptors (TLRs), important components of the innate immune system located on the epithelial surface, play major roles in tissue repair28). Among the known human TLRs, type 4 seems to play a crucial role in NEC development29). TLR4 may be activated by bacterial (i.e., lipopolysaccharides) or by other innate immune components. The activation of TLR4 signaling in the intestinal epithelium has been shown to inhibit enterocyte migration and to lead to enterocyte apoptosis in a mouse model, whereas inhibition of TLR4 signaling has been found to prevent NEC development and attenuate the degree of enterocyte apoptosis in mouse models and cell cultures27,28,29,30).
Developing fetuses express elevated TLR4 expression until the end of the gestation, owing to the fact that TLR4 regulates proliferation and differentiation of the intestinal epithelium. The persistently elevated expression of TLR4 during intrauterine life does not increase the risk of NEC for the fetus, probably because it lives in a sterile or quasi-sterile environment. At the end of gestation, the neonate shows low expression of TLR4, and the signal remains inactivated in the presence of a normal intestinal microflora. However, the expression of TRL4 in preterm babies is very high, and when the premature intestine is colonized by pathologic microflora, TLR4 signaling may become overactivated, leading to decreased ability of the body to repair the epithelium after injury28,29).
The effects of a TLR4-mediated response include gut barrier failure; bacterial translocation; intestinal inflammation; and, finally, a systemic inflammatory response through the activation of proinflammatory mediators such as IL-1, -6, -8, and -18; platelet-activating factor (PAF); leukotrienes; thromboxanes; free oxygen radicals; and nuclear factor kappa B (NFκB)4). Accumulation of ileal bile acids causes significant injury in the small intestine and acts in concert with the TLR4 pathway12). Intestinal integrity restitution requires intercellular connectivity, which is mediated through small channels (gap junctions), rich in connexin protein. Proinflammatory cytokines such as interferon gamma cause endocytosis of connexin (it is engulfed by the cell membrane and drawn in to the cell), thereby impairing intercellular connectivity and reducing the extent of intestinal restitution28).
Most premature infants do not develop NEC despite high TLR4 expression, which suggests that TLR4 signaling is weakened in the newborn intestinal epithelium by counter-regulatory mechanisms, including intra- and extracellular factors such as heat shock protein (Hsp), interleukin (IL) 1 receptor antagonist, IL-11 and -12, PAF acetylhydrolase, inhibitory protein of NF-κB, and EGF; thus, the development of NEC is limited. The intracellular factor Hsp70 (a major member of the Hsp family) is activated by stress, and plays a protective role in the intestine by limiting TLR4 signaling in enterocytes. TLR4 activation itself significantly increases Hsp70 expression in the enterocytes, which provides a mechanism of autoinhibition of the TLR4 signaling pathway. Reduced activity of Hsp70 or hyperactivation of TLR4 disrupts this balance and induces NEC, whereas upregulation of Hsp70 conversely leads to a reduction in TLR4 signaling31).
Furthermore, the extracellular factor EGF may also affect TLR4 signaling. The fetus continuously swallows amniotic fluid (extremely rich in EGF), which limits the amplification of TLR4 signaling in the fetal intestinal mucosa and in cultured enterocytes exposed to bacterial products, thus markedly reducing the degree of proinflammatory cytokine release. This extracellular factor inhibits TLR4 signaling via the peroxisome proliferator-activated receptor gamma and NF-κB pathways32).
3. Gut microbiota
Although many studies have report an association between NEC and infection, and despite the fact that many pathogens may simulate features of NEC, no specific microbe has been identified as a determinant etiologic factor33). Several findings33,34,35) such as the detection of abundant bacteria (including gram-negative pathogens) in the feces of NEC patients, a loss of gut microbial diversity and depletion of enterococcal populations in the feces before NEC development, and a correlation between the clinical finding of pneumatosis intestinalis and the presence of clostridial species (Clostridium butyricum and Clostridium paraputrificum) suggest that NEC may not result from a single causative species but from a microbial imbalance (dysbiosis), which may favor TLR4 activation and translocation of pathogens across the epithelium.
Future approaches to prediction
Identification of the infants at increased risk of NEC may be possible through methods currently available at a few research centers. These methods use noninvasive indicators such as profiling of the fecal microbiota36) and identification of the expression of inflammatory proteins from the buccal epithelium37). In addition, oxidative stress determined by measuring concentrations of nonprotein bound iron, advanced oxidation protein products, and total hydroxides in the cord blood has been reported to be a useful predictor of NEC in high-risk infants38). However, additional validation is required. Recently, Ng et al.39) reported that the levels of three intestinal biomarkers, namely liver-fatty acid binding protein (L), intestinal-fatty acid binding protein (I), and trefoil factor 3 (T), as well as the combined value of the three (LIT score), were significantly higher in patients with NEC than in those with septicemia or control groups. Furthermore, the expression of these biomarkers, including the LIT score, was also found to be higher in NEC nonsurvivors than in the survivors.
Newly emerging therapeutic strategies
There are many evidence-based approaches to prevent NEC (Table 2)40,41). These include withholding enteral feedings; using enteral antibiotics; feeding the infant with the mother's expressed breast milk; administering probiotic agents, prebiotic agents, or both; and administering various growth factors, anticytokine agents, and glucocorticoids. Recent advances in understanding the pathophysiology of NEC suggest that future treatments may involve immunological approaches such as pharmacologic inhibition of TLR signaling and manipulation of the intestinal environment other than by administration of specific nutrients. Some options have already been tested in clinical trials, and many others are likely to be developed and verified in a clinical setting in the future27).

Evidence of preventive measures for necrotizing enterocolitis
1. Nutritional support to preserve the epithelial barrier
Adequate feeding strategies and the use of specific molecules in enteral nutrition have been shown to have a protective effect on the risk of NEC. For example, human milk is able to protect against the development of NEC via induction of intestinal maturation and healing42). These positive effects of human milk have been attributed to several factors (i.e., macrophages, lymphocytes, secretory IgA, lysozymes, lactoferrin, oligosaccharides, nucleotides, cytokines, growth factors, PUFA, and enzymes) that influence host immunity, inflammation, and mucosal protection, but a specific component to transfer in preterm formula has not been definitively identified4,42).
Although several growth factors such as recombinant EGF, HB-EGF, and granulocyte colony-stimulating factor may play distinct and critical roles in normal intestinal development and repair following GI mucosal injury, further studies are needed to clarify the mechanisms of action of these growth factors43). Additionally, erythropoietin, erythropoietin-like growth factor, and cytokines may also play roles in the growth and development of the GI tract. In a rat hypoxia NEC model, pretreatment with erythropoietin resulted in a decrease in the incidence of NEC44).
There have been some studies comparing the incidence of NEC between infants breast-fed with fortified bovine milk-based or human milk-based supplements and those breast-fed without fortification. The Cochrane review by Kuschel and Harding45) presents the result of 13 randomized controlled trials that examined the long and short-term outcomes of preterm infants fed on a diet of fortified expressed breast milk. They found that the use of a fortifier was associated with improved growth, with no significant increase in the adverse outcomes, including NEC, in this group. In contrast, Sullivan et al.42) found a significantly higher incidence of NEC in infants receiving a bovine milk-based fortifier than in those receiving a human milk-based fortifier. However, the clinical significance of this result is questionable, as the incidence of NEC was small in both groups; moreover, it is possible that this finding may be the result of a dose-related association between increased expressed breast milk feeding and a reduced risk of NEC, rather than a true increase in risk due to the use of bovine milk fortifier46).
Endothelial nitric oxide, synthesized from the amino acid L-arginine, is an important regulator of vascular perfusion. Although the incidence of NEC has been found to be decreased with L-arginine supplementation in some studies, further human studies are needed47). Moreover, the use of zinc to prevent NEC is supported by evidences demonstrating the role of zinc in the maintenance of the epithelial barrier function and in the induction of adequate immune responses in experimental models of NEC; a recent clinical trial demonstrated that high-dose oral zinc supplementation decreased the incidence of NEC in preterm neonates48).
2. Modulation of inflammatory (TLR4) signal
The pharmacological upregulation of Hsp70 (inhibitor of TLR4 signal) within the intestinal mucosa by celastrol (a novel cell permeable triterpenoid antioxidant) has been tested in a mouse model of NEC49). In this model, celastrol was found to reduce enterocyte apoptosis and attenuate the severity of NEC, but studies in humans are still lacking. Hence, further studies are required for the establishment of these novel therapeutic options for the prevention and treatment of human cases of NEC.
Lipopolysaccharide (LPS), one of the most powerful proinflammatory bacterial virulence factors, directly induces TLR signaling. LPS-binding protein (LBP) is secreted by enterocytes in response to inflammatory stimuli and has concentration-dependent effects. At basal concentrations, LBP stimulates the inflammatory response by presenting LPS to its receptor; however, at high concentrations, LBP is able to neutralize LPS and prevent an exaggerated inflammatory response. Hence, targeting LBP may represent a novel therapeutic strategy to facilitate wound healing after the acute phase of NEC and other forms of intestinal injury26).
3. Modification of gut microbiota composition
Probiotic colonization improves GI health and prevents proinflammatory signaling and disease progression by increasing mucosal barrier function and by up-regulating the immune system. Additionally, it also reduces mucosal colonization of potential pathogens, and alters the key components of intestinal inflammation4). Several studies have demonstrated the efficacy and safety of prophylactic enteral probiotic administration in the prevention of NEC in infants with very low birth weight50,51,52,53,54,55,56). In these studies, the administered probiotics were Lactobacillus alone or in combination with Bifidobacterium (Table 3). Many aspects may have influenced the different results obtained in each trial. Importantly, the effect of probiotics has been shown to be dramatically decreased in areas where the occurrence of NEC is low (<5% of very low birth weight infants), suggesting that the baseline NEC rate seems to be a major factor affecting the potential benefit of probiotic supplementation in a specific population57).

Clinical trials for probiotic prophylaxis
In a recent Cochrane Database Systematic Review and meta-analysis published58), 16 randomized or quasi-randomized controlled trials that enrolled a total of 2,842 preterm infants at <37 weeks gestational age and/or with a birth weight of <2,500 g were evaluated. The included studies showed highly variable enrollment criteria (birth weight and gestational age), probiotic species, times of initiation, doses, formulations, durations of therapy of probiotics, and feeding methods. However, nonetheless, the authors concluded that enteral supplementation of probiotics significantly reduced the incidence of severe NEC (stage II or more) and mortality in preterm infants weighing <1,500 g at birth. On the other hand, there are currently insufficient data with regard to the benefits and potential adverse effects in the most atrisk infants, namely those weighing <1,000 g at birth.
U.S. Food and Drug Administration has not yet approved the use of probiotics in premature infants; however, this updated review of available evidence supports the routine use of probiotics in the neonatal intensive care unit58). Because probiotic products have not been subjected to rigorous manufacturing quality control, and because the contents of such products may not be reproducible in compliance with drug or pharmaceutical standards4), more comparative trials among probiotic products rather than placebo-controlled trials are needed to assess the most optimal preparation, dose, and duration for extremely low birth weight infants. The main limitation to future trials is the large sample sizes required to demonstrate the benefit of probiotics in this setting4,57,58).
Another proposed preventive strategy is to supplement with prebiotics, which enhance the growth of potentially beneficial intestinal microbes such as Bifidobacterium59). While the theoretical benefit has been reviewed, little information is currently available to support the benefit from their use in the prevention of NEC. Prebiotic agents include the oligosaccharides inulin, galactose, fructose, lactulose, and combinations of these nutrients.
4. Fecal transplantation
A novel perspective to manipulate intestinal microflora is represented by fecal transplantation. Fecal transplantation involves direct transfer of fecal material from a healthy donor to a recipient's upper or lower intestinal tract. This technique has proven effective in the treatment of refractory colitis by Clostridium difficile and in some cases of inflammatory bowel disease60). However, no data are currently available for neonates with NEC, and several important limitations should be considered before testing this therapeutic option in the newborn.
5. New surgical techniques
The most significant morbidity related to NEC is short bowel syndrome (SBS), which is an extensive, morbid condition with an increasing incidence. Currently, there are no functionally adequate surgical procedures for treating SBS. Bowel-lengthening procedures and parenteral and enteral nutrition have been shown to have modest effects. In a study from 1998, small bowel transplantation was reported to have a 5-year graft survival rate of 48%61), but this procedure is commonly accompanied by rejection and the need for lifelong immunosuppressant medications. Moreover, by serial transverse enteroplasty, a surgical method to increase the girth of the bowel in patients with SBS, the rate of independence from intravenous nutrition is only 58%62). Finally, tissue engineering techniques may represent a unique, novel strategy for intestinal failure after severe NEC in the future63).
Conclusions
NEC is one of the leading causes of mortality, and the most common reason for emergent GI surgery in newborns. However, NEC remains a major unsolved medical challenge, for which no specific therapy exists, and its pathogenesis remains controversial. A better understanding of the pathophysiology will offer new and innovative therapeutic approaches, and future studies should be focused on the roles of the epithelial barrier, innate immunity, and microbiota in this disorder. Bioinformatics modeling is a new emerging strategy aimed at understanding the dynamics of various inflammatory markers and their application in early diagnosis and treatment.
Notes
No potential conflict of interest relevant to this paper was reported.