Management issues of congenital adrenal hyperplasia during the transition from pediatric to adult care
Article information
Abstract
Steroid 21-hydroxylase deficiency is the most prevalent form of congenital adrenal hyperplasia (CAH), accounting for approximately 95% of cases. With the advent of newborn screening and hormone replacement therapy, most children with CAH survive into adulthood. Adolescents and adults with CAH experience a number of complications, including short stature, obesity, infertility, tumor, osteoporosis, and reduced quality of life. Transition from pediatric to adult care and management of long-term complications are challenging for both patients and health-care providers. Psychosocial issues frequently affect adherence to glucocorticoid treatment. Therefore, the safe transition of adolescents to adult care requires regular follow-up of patients by a multidisciplinary team including pediatric and adult endocrinologists. The major goals for management of adults with 21-hydroxylase deficiency are to minimize the long-term complications of glucocorticoid therapy, reduce hyperandrogenism, prevent adrenal or testicular adrenal rest tumors, maintain fertility, and improve quality of life. Optimized medical or surgical treatment strategies should be developed through coordinated care, both during transition periods and throughout patients' lifetimes. This review will summarize current knowledge on the management of adults with CAH, and suggested appropriate approaches to the transition from pediatric to adult care.
Introduction
Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive diseases caused by disorders in adrenal steroidogenesis1). Steroid 21-hydroxylase deficiency is the most common form of CAH, accounting for approximately 95% of cases2). Morbidities associated with CAH include adrenal insufficiency, genital ambiguity in females, and variable degrees of postnatal androgen excess2). With the advent of newborn screening and hormone replacement therapy, most children with CAH survive into adulthood. As a result, individuals with CAH who reach adolescence and adulthood can develop from several long-term complications, including short stature, precocious puberty, obesity, hypertension, polycystic ovary syndrome, infertility, tumor, osteoporosis, and reduced quality of life234).
Management of CAH during adolescence and the transition from pediatric to adult care are challenging tasks because hormonal changes during puberty can lead to inadequate suppression of adrenal androgens3). In addition, psychosocial status affects adherence to medication in adolescence and young adulthood4). Therefore, treatment regimens should be reassessed during adolescence and adulthood. Successful transition from pediatric to adult care requires regular follow-up of patients by a multidisciplinary team including pediatric endocrinologists, urologists, gynecologists, psychiatrists, and adult endocrinologists. The transition process should be prepared and planned gradually in order to empower the adolescent to assume greater responsibility for their own health-care3). Thus, thorough education of patients and health-care providers is critical for successful transition from pediatric to adult care.
The major treatment goals during transition from pediatric to adult care are to minimize the long-term complications of glucocorticoid therapy, to reduce adrenal hyperandrogenism, to prevent adrenal or testicular adrenal rest tumors (TARTs), to maintain fertility, and to optimize quality of life3). New medical and surgical treatment strategies, transition protocols, and coordinated care systems are being developed to improve the long-term outcomes of adults with CAH3). However, it is imperative that health-care providers perform further research to investigate the natural history of CAH in adults, and to establish optimal interventions for effective transition programs that will improve treatment outcomes.
This review describes current knowledge regarding the long-term consequences of CAH in adolescence and adulthood and management issues during the transition period from child to adult care.
Hormonal changes in patients with 21-hydroxylase deficiency during puberty
Profound hormonal changes occur during the transition from childhood to adulthood. Pubertal activation of the hypothalamic-pituitary-gonadal axis and increases in growth hormone and insulin-like growth factor 1 secretion have been shown to decrease 11β-hydroxysteroid dehydrogenase type 1 activity and increase glomerular filtration rate, resulting in increased cortisol clearance45). These changes can decrease the effectiveness of glucocorticoid drugs. In addition, age-related physiological increases in adrenal androgens and insulin resistance are amplified in adolescents with 21-hydroxylase deficiency, which can complicate management of the condition4).
Long-term consequences of 21-hydroxylase deficiency in adolescents and adults
Adolescents and adults with 21-hydroxylase deficiency can experience long-term complications such as short stature, precocious puberty, obesity, hypertension, hirsutism, polycystic ovary syndrome, adrenal or testicular tumors, infertility, and osteoporosis36).
Linear growth is initially accelerated in children with CAH; however, final adult height is reported as one to 2 standard deviations below the mean for patients' target heights (Table 1)7). Inadequately treated patients may not reach their potential target height because adrenal androgens cause premature epiphyseal fusion. Overtreatment with glucocorticoids is also strongly associated with poor height outcomes8). Therefore, it is important to use the lowest possible glucocorticoid doses in order to optimize final adult height8).

Auxological characteristics of adults with congenital adrenal hyperplasia caused by 21-hydroxylase deficiency
Obesity is common in patients with CAH, and children with 21-hydroxylase deficiency have a high body mass index (BMI) throughout childhood and adulthood (Table 1)9). These data suggest that many patients with 21-hydroxylase deficiency are overtreated10). In previous reports, children with CAH had a higher BMI in 16%–25% of cases1112). In addition, hypertension is prevalent in children and adolescents with CAH and has been related to BMI independent of glucocorticoid or mineralocorticoid therapy13). Lifestyle interventions are recommended to prevent the potential metabolic consequences of obesity2).
Common tumors in adults with 21-hydroxylase deficiency include TARTs and tumors of the adrenal cortex. Adrenal masses occur in 1%–4% of general population14). High prevalence of adrenal tumors has been observed in adults with CAH, especially among those with inadequate glucocorticoid therapy15). The enlargement of the adrenal glands in 21-hydroxylase deficiency predisposes sufferers to adrenal tumors, including myelolipomas16). Myelolipomas are commonly found in patients with 21-hydroxylase deficiency and can be enormous, requiring removal for mass effect16). However, it is recommended that adrenal imaging is reserved for patients manifesting atypical clinical courses or biochemical findings2).
TARTs are one of the most common complications in male CAH patients with insufficient glucocorticoid replacement therapy6). Around 30%–50% of men with 21-hydroxylase deficiency develop TARTs17). These TARTs are benign and decrease in size after high doses of glucocorticoid therapy1819). If TARTs are unresponsive to glucocorticoid therapy, surgical intervention using a testis-sparing procedure and cryopreservation of the semen may be necessary1720). It has been recommended that periodic ultrasonography is performed to evaluate TARTs during follow-up of males with CAH2).
CAH has implications for reproductive functions such as menstruation and fertility. Irregular menstrual cycles commonly occur in females with CAH who receive inadequate treatment21). Females with poorly-controlled classic CAH can also experience hirsutism, oligomenorrhea, amenorrhea, menorrhagia, and acne; a collection of symptoms often observed in polycystic ovary syndrome8). Functional ovarian hyperandrogenism can cause irregular menstruation even in women with well-controlled CAH22). However, routine pelvic ultrasonography is not required for females with CAH who have regular menstrual cycles2).
Impaired fertility is reported in both males and females with 21-hydroxylase deficiency. Affected women experience a number of impediments to fertility, including vaginal insufficiency with dyspareunia, anovulation, and polycystic ovary syndrome caused by elevated adrenal androgens3). Reduced fertility is especially common among those with salt-losing forms of CAH2023) due to high adrenal androgen levels that lead to anovulatory cycles24), and incomplete surgical reconstruction impairing sexual intercourse25). Fertility rates in simple virilizing CAH have been reported to be around 50%23). In women with nonclassic CAH, a retrospective multicenter review demonstrated that 68% of 101 women had become pregnant before the diagnosis was made, implying that glucocorticoid treatment is not essential for fertility26). In males with 21-hydroxylase deficiency, the mechanism of impaired fertility is gonadotropin suppression by adrenal androgens when adequate doses of glucocorticoids are not administered, which results in testicular atrophy impairing spermatogenesis2728). In addition, the development of TARTs cause obstruction of the seminiferous tubules and the normal testis tissue, leading to impaired blood flow, functional impairment, and fibrosis3). In a large cohort study, TARTs were found in 34% of patients, and severe oligospermia and azoospermia were prevalent in 70% of these cases29).
A major complications of long-term glucocorticoid treatment is osteoporosis. Glucocorticoid therapy inhibits osteoblastic activity, leading to decreased bone mineral density30). Reduced bone mineral density has been inversely correlated with cumulative doses of glucocorticoid31). A retrospective study of 62 adult women with CAH found that overtreatment with glucocorticoids lead to osteoporosis, along with an increased incidence of fractures32). However, routine bone mineral densitometry is not recommended because there is no evidence of an increased incidence of osteoporosis among adults with CAH who do not receive overtreatment3334). Further studies into longitudinal follow-up of bone mineral density would be useful to investigate the occurrence of osteoporosis in adult patients with 21-hydroxylase deficiency2).
Assessment of adolescents and adults during transition from pediatric care
Prevention and management of long-term complications, and maintaining quality of life is a major concern for adolescents and adults with 21-hydroxylase deficiency3). Patient medical histories and physical signs of overtreatment or undertreatment for CAH should be considered. This could include reviewing past treatment regimens, frequency of adrenal crises, adherence to medication, regular menstruation, cushinoid features, skin hyperpigmentation, and testicular size3). Extensive education of patients and coordination of services among health-care providers for adults with 21-hydroxylase deficiency remains a major task during the transition from pediatric to adult care35). The patient's knowledge of the disease should be evaluated, as well as understanding of stress dose of glucocorticoids and emergency use of hydrocortisone3).
Medical treatment of adolescents and adults with 21-hydroxylase deficiency
In children with CAH, glucocorticoid doses should be adjusted to prevent hyperandrogenism and optimize growth. Glucocorticoid must be given in doses sufficient to suppress adrenal androgen secretion, without total suppression of the hypothalamic-pituitary-adrenal axis2). The recommended dose of hydrocortisone for growing children is 10–15 mg/m2/day2). In adults with CAH whose growth and development are complete, the goal of treatment is to prevent long-term complications, to preserve fertility and satisfactory sexual function, and to maintain general wellbeing and physical performance6). Pediatric doses of glucocorticoids should be reassessed during the transition to adult care in order to avoid overtreatment. High dose glucocorticoid therapy can lead to cushinoid features, short stature, weight gain, hypertension, hirsutism, infertility, osteoporosis, and impaired glucose tolerance6), while excess mineralocorticoids may result in hypertension2). Adults with classic CAH are treated with physiologic doses of hydrocortisone to prevent adrenal insufficiency; however, this does not suppress adrenocorticotropic hormone and adrenal androgen production. Inadequate androgen suppression in CAH can result in short stature, early puberty, virilization in females, infertility, adrenal tumors, TARTs in males, and adverse psychological sequelae4). As a result of these factors, it is difficult to suppress adrenal androgen secretion without overtreatment with glucocorticoid, and patients often experience hypercortisolism, androgen excess, or a combination of these states (Fig. 1)9).
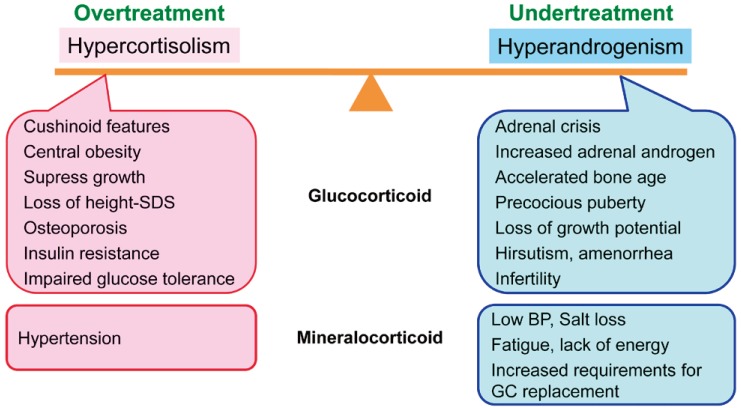
Balance between overtreatment and undertreatment in the management of adults with 21-hydroxylase deficiency. If a glucocorticoid is administered at physiologic dose to prevent adrenal insufficiency, hyperandrogenism persists. However, if the glucocorticoid dose is increased to suppress excess adrenal androgen, hypercortisolism can occur. SDS, standard deviation score; BP, blood pressure; GC, glucocorticoid.
Hydrocortisone (15–25 mg/day) is the first line of therapy for 21-hydroxylase deficiency. However, 3 times daily medication is required due to its short half-life, and this can lead to poor treatment adherence3). The second line of therapy is to add a small bedtime dose of a long-acting glucocorticoids, such as prednisolone (1–2 mg) or dexamethasone (0.1–0.5 mg)3). If this regimen is insufficient to achieve control of the condition, or represents too many doses during the day, twice daily prednisolone (4–6 mg/day) is the third alternative2). The fourth line of treatment consists of once or twice daily dexamethasone (0.25–0.5 mg/day), although this should be used cautiously because of the associated complications, such as iatrogenic Cushing syndrome3).
The need for mineralocorticoids decreases with age, because levels of serum aldosterone are high and renal mineralocorticoid receptor mRNA are low at birth36). Doses of fludrocortisone can be substantially reduced in adults, and may no longer be required in some patients, even those with salt-losing forms of CAH37). However, optimal dosage of fludrocortisone in adults has not been studied, and the adequacy of mineralocorticoid supplementation must be monitored by observing blood pressure and plasma renin activity2).
Laboratory testing and monitoring of adolescents and adults with 21-hydroxylase deficiency
Regular appropriate hormone measurements should be taken during the transition from pediatric to adult care. Serum 17α-hydroxyprogesterone (17-OHP) is the standard for diagnosis and a sensitive parameter for assessing disease control; however, correlation of 17-OHP with androgen levels in adults is highly variable3). Levels of sex hormone binding globulin and total testosterone levels can be used to assess to calculate free testosterone in women with 21-hydroxylase deficiency3). Follicular-phase serum progesterone levels should be assessed in women with chronic amenorrhea and be maintained below 0.6 ng/mL38). In males with 21-hydroxylase deficiency, androstenedione and gonadotropin levels should be checked in order to monitor adrenal androgens and testicular function3). Periodic screening for TARTs by testicular ultrasonography is also necessary from adolescence onward2).
Treatment of adults with nonclassic CAH
In cases where enzyme activity is preserved to a certain degree, patients with 21-hydroxylase deficiency remain asymptomatic until puberty6). Most patients with the nonclassic form present with hirsutism or menstrual irregularities at the time of puberty or early adolescence6). Asymptomatic adults do not require daily glucocorticoid treatment. However, it is recommended that children with inappropriately early onset and rapid progression of pubarche and adolescents with overt virilization should be treated2).
Management of women with 21-hydroxylase deficiency during pregnancy
For pregnant women who experience difficulties due to vaginal stenosis or dyspareunia, vaginal reconstruction surgery can be performed. However, some women suffer bladder infections or abscesses, cysts, or scar tissue35). Women with CAH should be managed by both endocrinologists and obstetricians during pregnancy2). Pregnant women with 21-hydroxylase deficiency should be treated with hydrocortisone or prednisolone239). Dexamethasone should not be used to treat the pregnant woman with CAH because it is not inactivated by placental 11β-hydroxysteroid dehydrogenase type 2, and thus can suppress the fetal adrenal glands and cause low birth weight240). In some cases, slightly higher doses of glucocorticoid may be needed to induce ovulation. In such cases, serum progesterone levels should be maintained below 0.6 ng/mL38). In the third trimester of pregnancy, glucocorticoid dose may be increased up to 50%37). During labor and delivery, stress doses of glucocorticoids should be administered; however, there are no controlled studies or guidelines regarding optimal doses of glucocorticoids at this point2).
Psychological support and counseling
Exposure to excess androgens during the prenatal period influences brain development of females with classic CAH41). Females with classic CAH may display more sexual concerns and masculine behavior than unaffected females424344). They are less likely to have sexual relations than females in control groups, and those with the salt-losing form have poorer psychosocial functioning45). Although gender-atypical behavior has been reported, women with CAH frequently identify themselves as female and reversal of gender identity is extremely rare4144). In contrast, severely virilized females may be raised as males initially, and once such an assignment made, it can be difficult to reverse gender identity2).
There is a lack of guidelines regarding psychosexual management of patients with CAH. Patients may have psychosocial problems associated with disorders of sexual development and need specialized care by psychiatrists with expertise in such problems2). Multidisciplinary guidance in the area of psychosexual function is critical with regards to gender identity or dysphoria6).
Multidisciplinary team approach during transition period
Unfortunately, a considerable number of CAH patients are lost to regular follow-up once they move out of pediatric care6). This is likely the result of inadequate cooperation between pediatric and adult endocrinologists at the time of transition to adult care. This transition should be processed gradually46), therefore, it is recommended that pediatric endocrinologists, gynecologists, urologists, psychiatrist, and adult endocrinologists perform joint clinics for transferring CAH patients to adult care2). Adults with 21-hydroxylase deficiency require careful screening for tumors, osteoporosis, obesity, and maintenance of general health35). The approach to adults with 21-hydroxylase deficiency requires detailed information on the patient's prior treatment, the natural history and long-term consequences of the disease, and the influence of various treatments on adrenal and gonadal steroidogenesis. Therefore, it would be desirable to have adult endocrinologists with special expertise in the treatment of CAH patients6).
Conclusions
Patients with CAH must experience a transition from pediatric to adult care, and can develop various long-term complications. Transition of adolescents to adult care involves changes in treatment goals toward prevention of long-term complications and optimization of quality of life. Careful monitoring, extensive education, and follow-up should be required for patients with CAH, with greater vigilance in the monitoring of biochemical and clinical features. Optimal treatment of adults with 21-hydroxylase deficiency can be achieved through a multidisciplinary approach, including consultation with pediatric and adult endocrinologists. Further investigations are needed to investigate the natural history of CAH and to develop management guidelines for adolescents and adults with 21-hydroxylase deficiency.
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.
