Application of array comparative genomic hybridization in Korean children under 6 years old with global developmental delay
Article information
Abstract
Purpose
Recent advancements in molecular techniques have greatly contributed to the discovery of genetic causes of unexplained developmental delay. Here, we describe the results of array comparative genomic hybridization (CGH) and the clinical features of 27 patients with global developmental delay.
Methods
We included 27 children who fulfilled the following criteria: Korean children under 6 years with global developmental delay; children who had at least one or more physical or neurological problem other than global developmental delay; and patients in whom both array CGH and G-banded karyotyping tests were performed.
Results
Fifteen male and 12 female patients with a mean age of 29.3±17.6 months were included. The most common physical and neurological abnormalities were facial dysmorphism (n=16), epilepsy (n=7), and hypotonia (n=7). Pathogenic copy number variation results were observed in 4 patients (14.8%): 18.73 Mb dup(2)(p24.2p25.3) and 1.62 Mb del(20p13) (patient 1); 22.31 Mb dup(2) (p22.3p25.1) and 4.01 Mb dup(2)(p21p22.1) (patient 2); 12.08 Mb del(4)(q22.1q24) (patient 3); and 1.19 Mb del(1)(q21.1) (patient 4). One patient (3.7%) displayed a variant of uncertain significance. Four patients (14.8%) displayed discordance between G-banded karyotyping and array CGH results. Among patients with normal array CGH results, 4 (16%) revealed brain anomalies such as schizencephaly and hydranencephaly. One patient was diagnosed with Rett syndrome and one with Möbius syndrome.
Conclusion
As chromosomal microarray can elucidate the cause of previously unexplained developmental delay, it should be considered as a first-tier cytogenetic diagnostic test for children with unexplained developmental delay.
Introduction
Global developmental delay is defined as significant delay (2 or more standard deviations) in two or more developmental domains: gross or fine motor skills, speech and language, cognition, personal and social interactions, and activities of daily living12). The term ‘global developmental delay’ is usually reserved for younger children (i.e., typically less than 5 years of age) while the term ‘intellectual disability’ is usually applied to older children when intelligence quotient testing is valid and reliable3). Global developmental delay is estimated to affect 1%–3% of children, many of whom will display intellectual disability with older age12).
It is difficult to make an etiologic diagnosis for patients with global developmental delay because they do not reveal sufficient specific medical history or clinical features. Recent introduction of chromosomal microarray analysis such as array comparative genomic hybridization (CGH) and single nucleotide polymorphism array has greatly contributed to the discovery of genetic causes of unexplained developmental delay45). CGH enables more sensitive detection of DNA copy-number changes on a genome-wide scale with at least 10x higher resolution compared to that of conventional karyotyping4). Chromosomal microarray has been recently recommended as a first-tier cytogenetic diagnostic test for patients with developmental delay or congenital anomalies5). However, although chromosomal microarray analysis is widely used for research purposes, its use is still not permitted in the field of medical practice in Korea.
Here we aimed to describe the results of array CGH and clinical features in 27 Korean children with global developmental delay accompanied by physical or neurological problems. In addition, we summarized the phenotypes of five cases showing abnormal or uncertain significant results of array CGH in conjunction with the genetic testing results.
Materials and methods
1. Patients
We included 27 patients who fulfilled the following conditions for this study: (1) Korean children under 6 years with global developmental delay who were admitted to the Department of Pediatrics at Ulsan University Hospital between April 2011 and February 2014; (2) children who had at least one more physical or neurological problems other than global developmental delay, such as facial dysmorphism, polydactyly, epilepsy, and hypotonia; and (3) patients in whom both array CGH and G-band karyotyping tests were performed. We retrospectively reviewed the medical records of those patients to obtain their clinical information. Written informed consent was obtained from all patients' parents or guardians. This study was approved by the Institutional Review Board of Ulsan University Hospital (IRB No. 2011-04-038).
2. Array CGH and confirmatory tests
Oligonucleotide-based microarray analysis was performed using a 135K CGX-3 whole-genome microarray platform (Roche NimbleGen Inc., Madison, WI, USA). Genomic DNA was extracted from the patients' peripheral blood samples. Patients' DNA and normal control DNA (Human Genomic DNA: Male/Female, Promega, Madison, WI, USA) were then labeled with Cy3 and Cy5 dyes via NimbleGen Dual-Color DNA Labeling Kit (Roche NimbleGen Inc.). Array hybridization and washing were performed according to the manufacturer's instruction. The array was scanned at a resolution of 2.5 µm using NimbleGen MS 200 Microarray Scanner. Results were then analyzed using NimbleScan v2.6 software (Roche NimbleGen Inc.) and signal intensity ratios were analyzed using custom oligonucleotide array CGH analysis software (Genoglyphix, Signature Genomics, Spokane, WA, USA) with the use of aberration filter of 3 contiguous probes. The criteria for quality control metrics were determined using standard deviation less than 0.14 and median absolute deviation less than 0.23 as major parameters for high-quality array reference values. Log2 values less than 0.3 were excluded from the analysis, and the patient to control ratio of 2:3 or greater were considered to be copy number variations (CNVs). Public databases, such as the American College of Medical Genetics Practice Guidelines, University of California Santa Cruz Genome Browser (http://genome.ucsc.edu), Decipher (http://decipher.sanger.ac.uk), the Database of Genomic Variants (http://projects.tcag.ca/variation/), the International Standards for Cytogenomic Arrays (http://iscaconsortium.org), the National Center for Biotechnology Information, and the Genoglyphix Chromosome Aberration Database, a genoglyphix-user database provided by genolgyphix, were used to classify and interpret the identified CNVs into benign CNVs, pathogenic CNVs or variants of uncertain significance. All pathogenic CNVs and variants of uncertain significance detected by microarray analysis were confirmed by fluorescence in situ hybridization (FISH) or real-time quantitative polymerase chain reaction (qPCR). FISH was performed using bacterial artificial chromosome probes located in the regions of gain or loss (target region) and other regions (internal control). DNA of bacterial artificial chromosome clones, RP11-480N14 (2p24.3), RP11-314N13 (20p13), RP11-20A12 (2p21), RP11-265P9 (2p22.3), RP11-141C15 (2q22.3), RP11-1069N19 (4q22.3), and RP11-241P10 (4p16.3) (BACPAC Resources Center, Children's Hospital Oakland Research Institute, Oakland, CA, USA), were labeled by nick translation (Abbott Molecular Inc., Des Plaines, IL, USA) with Enzo Red-dUTP or Enzo Green-dUTP (Abbott Molecular Inc.). A minimum of 150 metaphase and 50 interphase cells were counted to determine the percentage of mosaicism in Phytohaemagglutinin-stimulated cell cultures. qPCR was performed using SensiFAST SYBR Hi-ROX Kit (Bioline, London, UK) on a StepOne Real-Time PCR System apparatus (Applied Biosystem, Foster City, CA, USA). Each sample was analyzed in triplicate for target gene and reference gene. In each assay, a normal control and no template control were included. The relative gene copy number was quantified by the comparative CT method6). The costs of array CGH and confirmatory tests, such as FISH and qPCR were not charged to the patients.
3. Other diagnostic tests
To make etiologic diagnosis of global developmental delay accompanied with physical or neurological abnormality, the following tests were also selectively performed before, after or simultaneously with array CGH and G-banded karyotyping: (1) magnetic resonance imaging (MRI) of the brain; (2) genetic testing for the following known genetic disorders: fragile X syndrome, Prader-Willi/Angelman syndrome, Rett syndrome, spinal muscular atrophy, myotonic dystrophy, DiGeorge syndrome, Williams syndrome, and infantile neuronal ceroid lipofuscinosis; (3) workup for metabolic disorders, such as plasma amino acid analysis, urine organic acid analysis, tandem mass spectrometry, electron microscopic study on skin for neuronal ceroid lipofuscinosis, and Berry spot test and cetylpyridinium chloride precipitation test for mucopolysaccharidosis, and (4) neurophysiologic studies such as electroencephalography, electromyography and nerve conduction study, brainstem auditory evoked potential, and visual evoked potential.
Results
1. Clinical characteristics of patients
Fifteen male patients and 12 female patients with a mean age of 29.3±17.6 months (range, 6–65 months) were included in this study. The number of patients according to their age was 7 (6–12 months), 6 (1–2 years), 4 (2–3 years), 5 (3–4 years), 4 (4–5 years), and 1 (5–6 years). The most common physical and neurological abnormalities other than global developmental delay which patients revealed were facial dysmorphism (n=16), epilepsy (n=7), and hypotonia (n=7). Other physical and neurological problems included brain anomalies such as schizencephaly and hydranencephaly (n=4), polydactyly (n=3), short and thick fingers (n=3), visual disturbance (n=2), failure to thrive (n=2), strabismus (n=2), flat feet (n=2), simian crease (n=2), syndactyly (n=1), multicystic kidney (n=1), ataxic gait (n=1), dystonia (n=1), macrocephaly (n=1), microcephaly (n=1), facial asymmetry (n=1), sensory neuronal hearing loss (n=1), aortic valve stenosis (n=1), hepatomegaly (n=1), and expressionless face (n=1). All of the patients' parents are Koreans and nonconsanguineous.
2. Result of array CGH and G-banded karyotyping
Four patients (14.8%) revealed 6 pathogenic CNVs in array CGH. Three CNVs were deletions and the remaining three were duplications. The size of the 6 pathogenic CNVs ranged from 1.19 Mb to 22.31 Mb. There were no CNVs associated with specific known syndromes. One patient (3.7%) showed a CNV of uncertain significance (461.17-kb deletion). The clinical phenotypes and genetic testing results of the 5 patients who showed pathogenic CNVs and variants of uncertain significance in array CGH are described in Table 1.

Clinical and genetic features in 5 patients who showed pathogenic copy number variations and variants of uncertain significance in array comparative genomic hybridization
Three patients (11.1%, patients 1, 2, and 3) revealed abnormalities in G-banded karyotyping. These 3 patients also showed evidence of pathogenic CNVs in array CGH. Aberrations detected by karyotyping were larger than 12 Mb on array CGH. Four patients (patients 1, 2, 4, and 5) displayed discordance between G-banded karyotyping and array CGH results. Patient 1 showed 46,XY,dup(20)(p13) in G-banded karyotyping, and an 18.73 Mb duplication of 2p24.2-2p25.3 and 1.62 Mb deletion of 20p13 in array CGH. The results were interpreted as a rearrangement that resulted from duplication of the 2p terminal adhered to the deleted 20p terminal, which was then verified by FISH (Fig. 1). Patient 2 had low-level mosaicism (11.5%) of 46,XY,dup(2)(p21p25.1[177]/46,XY,del(2)(p22p23)[23]) in G-banded karyotyping, and a 22.31 Mb duplication of 2p22.3-2p25.1 and 4.01 Mb duplication of 2p21-2p22.1 in array CGH. In patient 2, array CGH analysis did not show low-level mosaicism, but was able to identify that the one large duplication of dup(2) (p21p25.1) observed in G-banded karyotyping was composed of 2 separate duplications (2p21-2p22.1 and 2p22.3-2p25.1) containing genome of normal copy number between them, which was also verified by FISH (Fig. 2). Patients 4 and 5 revealed normal G-banded karyotype, but demonstrated a 1.19 Mb and 461 Kb deletions, respectively, in array CGH. Patient 1 also harbored a duplication of the MYCN locus at 2p24.3, which is known to be responsible for the development of neuroblastoma78). However, careful work-up for neuroblastoma at 32 months of age revealed no sign of the disease.
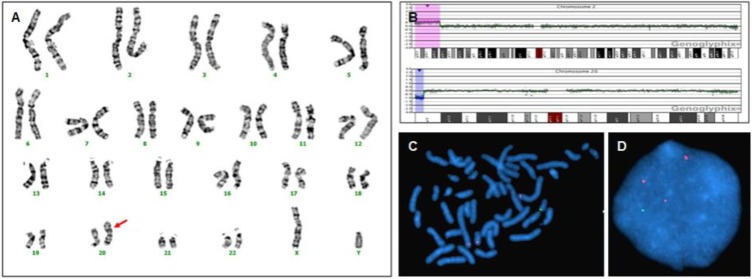
Genetic features of patient 1. (A) G-banded karyotyping shows 46,XY,dup(20)(p13) (arrow). (B) Array comparative genomic hybridization shows an 18.73-Mb duplication of 2p24.2-2p25.3 (red region) and 1.62-Mb deletion of 20p13 (blue region), (C, D) Fluorescence in situ hybridization indicates a rearrangement resulting from fusion of the duplicated 2p terminal to the deleted 20p terminal. Bacterial artificial chromosome (BAC) RP11-480N14 (red) harbors the 2p24.3 location and BAC RP11-314N13 (green) harbors the 20p13 location.
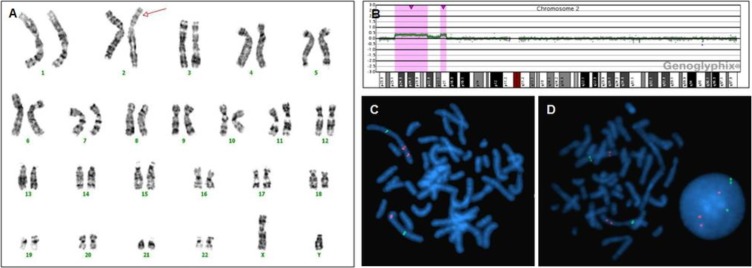
Genetic features of patient 2. (A) G-banded karyotyping shows 46,XY,dup(2)(p21p25.1) in this panel (arrow). However, patient 2 had low-level mosaicism (11.5%) of 46,XY,dup(2)(p21p25.1[177]/46,XY,del(2)(p22p23)[23]) in G-banded karyotyping. (B) Array comparative genomic hybridization (CGH) shows a 22.31-Mb duplication of 2p22.3-2p25.1 and 4.01-Mb duplication of 2p21-2p22.1 (red regions). Array CGH analysis did not show low-level mosaicism, but was able to identify that the large duplication of dup(2)(p21p25.1) observed in G-banded karyotyping was actually composed of two separate duplications (2p21-2p22.1 and 2p22.3-2p25.1), whereas the copy number of the intermediate genomic sequence was normal. (C) Fluorescence in situ hybridization (FISH) indicates 2p24.3 duplication (RP11-480N14, red color), and normal copy number of 2q22.3 (RP11-141C15, green color). (D) FISH also indicates 2p21 duplication (RP11-20A12, red color).
Among the 5 patients with pathogenic CNVs and variants of uncertain significance in array CGH, only one patient's parents (patient 2) underwent the G-banded karyotype test, the results of which were normal. No patients' parents underwent the other genetic testing including array CGH. The parental phenotypes of all 5 patients who showed pathogenic CNVs and variants of uncertain significance were discordant to the probands. All 27 patients had benign CNV loci, which were composed of losses of 42 loci in 23 patients and gains of 32 loci in 19 patients.
3. Results of other diagnostic tests
Diagnostic tests other than array CGH and G-banded karyotyping performed in the 27 patients are described in Table 2. Twenty-five patients (92.6%) underwent MRI of the brain, which was the most commonly performed diagnostic examination other than array CGH and G-banded karyotyping. Four patients (16%) revealed brain anomalies that were recognized as the causes of developmental delay such as schizencephaly, hydranencephaly, pachygyria in both frontal and temporal lobes, and subependymal heterotopia. All 4 patients with brain anomalies displayed normal result in array CGH and G-banded karyotyping tests despite accompanying physical problems such as facial dysmorphism, polydactyly, or hypotonia. Genetic testing for 8 known genetic disorders was performed in 17 patients (63.0%). Only 1 patient (5.9%) was diagnosed with Rett syndrome from the result of the MECP2 gene test. Diagnostic tests for metabolic disorders were performed in 14 patients (51.9%). No patient was diagnosed with specific metabolic disorder. Neurophysiologic studies were performed in 16 patients (59.2%). Ten of 11 patients (90.9%) who underwent electroencephalography displayed abnormal findings, including 2 patients with focal epileptiform discharges, 2 patients with background abnormality, and 6 patients with both abnormalities. Seven of these patients were also diagnosed with epilepsy. Nerve conduction study of the face revealed abnormal findings in 1 patient with facial asymmetry. Two of 10 patients (20 %) who underwent brainstem auditory evoked potential displayed abnormal results. No etiologic diagnosis of global developmental delay was made by the neurophysiologic study despite abnormal results. Finally, one patient was also diagnosed with Möbius syndrome by the clinical features such as facial paralysis and strabismus.

Tests other than array comparative genomic hybridization and G-banded karyotyping performed in 27 patients with global developmental delay accompanied with physical or neurological problems
Discussion
In the present study, four of 27 patients (14.8%) revealed 6 pathogenic CNVs, and 1 patient (3.7%) revealed a variant of uncertain significance in array CGH analysis, whereas 3 patients (11.1%) revealed abnormalities in G-banded karyotyping. Based on the number of patients with pathogenic CNVs divided by the total number of patients tested, there was no significant difference of diagnostic yield between array CGH and G-banded karyotyping in this study (14.8% vs. 11.1%). However, four of 5 patients showing pathogenic CNVs and variants of uncertain significance in array CGH demonstrated discordant results compared to those of G-banded karyotyping. Whereas G-banded karyotyping identified only large-sized genomic imbalances, array CGH analysis was able to detect additional genomic imbalances smaller than 5 Mb. Moreover, array CGH analysis could provide the exact location and size of the genomic imbalance, unlike G-banded karyotyping, which also enabled analysis of whether specific known genes were involved in the abnormalities.
Before microarray technology was available, tests such as G-banded karyotyping, FMR1 gene testing, subtelomeric FISH, MeCP2 gene testing, MRI of the brain, and visual and hearing assessments were recommended for evaluation of unexplained global developmental delay based on a diagnostic yield of greater than 1%9). Diagnostic yield of chromosomal microarray analysis in children with developmental delay/intellectual disability has been reported between 7% and 20%101112131415161718). One literature review regarding the use of chromosomal microarray as a diagnostic test for patients with developmental disabilities, autism, or congenital abnormalities reviewed 33 original reports, including 21,698 patients tested by chromosomal microarray, showed that the average detection rate of pathogenic CNVs was 12.2%, whereas the average diagnostic yield of G-banded karyotyping was less than 3% if Down syndrome was excluded5). Genome-wide array (~100 kb) showed diagnostic yield of 11%–15%, whereas targeted chromosomal microarray showed diagnostic yields of 7%–11%5). In another literature review, in which 367 research articles were selected through literature searches for the years 1980–2009, revealed that the average diagnostic yield of chromosomal microarray in subjects with unexplained global developmental delay/intellectual disability was 7.8% with a range of 0%–50%, whereas those of karyotype studies and FISH were 4% and 3.5%, respectively19). As patients with multiple CNVs usually present with a more severe phenotype than a single CNV20), identification of additional CNVs even in patients who had chromosomal aberrations already detected in karyotyping is useful for not only genetic counseling, but also in understanding the phenotype of the patients.
Diagnostic yield has increased as the resolution of cytogenetic tests for patients with developmental delay has improved. Although conventional karyotyping can detect chromosomal abnormalities larger than 5 Mb, genomic imbalances in the range of 5–10 Mb are often undetected. However, the development of array CGH technology has enabled the detection of genomic imbalances of less than 500 kb45). Resolution of array CGH is determined by the density of coverage across the genome (spacing) and size of nucleic acid targets (length of DNA probe)21). Uniform coverage throughout the genome with resolution less than 400 kb is usually recommended as a chromosomal microarray platform, although chromosomal microarray analysis with much higher resolution is commercially available nowadays5). This study also showed that four CNVs less than 5 Mb which could not be detected in conventional karyotyping were detected by array CGH.
In this study, we included children who had not only global developmental delay but also physical or neurological abnormalities with the expectation of higher diagnostic yield of array CGH compared to that in children with developmental delay only. Abovementioned large-scale literature review demonstrated higher average diagnostic yield of chromosomal microarray in subjects with syndromic features than in those with global developmental delay/intellectual disability (10.6% vs. 7.8%)19). Diagnostic yield of the pathogenic CNVs in the present study was 14.8%. However, since this is a small case series study, it could not be assessed whether the results indicate that array CGH analysis in patients with global developmental delay accompanied with physical or neurological abnormalities offers higher yield than in patients with developmental delay only.
In regard to association of severity of developmental delay/intellectual disability with diagnostic yield of array CGH, a recent report, which included 329 children with developmental delay/intellectual disability, demonstrated 16% diagnostic yield of array CGH without difference of diagnostic yield according to the degree of developmental delay/intellectual disability. Based on their results, the authors emphasized the need to conduct array CGH on all patients with developmental delay/intellectual disability, regardless of the severity22).
Although array CGH analysis can identify aneuploidies, microdeletion or microduplication, and unbalanced chromosomal rearrangement on a genome-wide scale with high resolution, it may not detect balanced translocation or inversion, polyploidy, and mosaicism45). To identify chromosomal aberrations that may go undetected in array CGH, G-banded karyotyping should be conducted together with array CGH analysis in cytogenetic testing. However, only a small portion (<1%) of all cases of unexplained developmental delay/intellectual disability and/or congenital anomalies reveal balanced rearrangement and low-level mosaicism523). In the present study, low-level mosaicism (11.5%) in 1 patient was detected by karyotyping, but not by array CGH. This suggests that G-banded karyotyping can play a role in the genetic diagnosis of developmental delay despite advances in chromosomal microarray testing.
Clinical interpretation of CNVs detected in array CGH is critical and often very complicated. CNVs are classified into pathogenic, benign, and variants of uncertain significance. CNVs present in databases of healthy individuals, CNVs associated with well-defined clinical phenotype/syndrome, identical CNVs inherited from an affected parent, expanded CNVs, especially deletions containing many genes, or morbid genes reported in the Online Mendelian Inheritance in Man can be easily categorized into benign and pathogenic5). However, since enormous genetic variations are present among healthy individuals and populations, categorization of CNVs is often a tough process. Recent data on CNVs detected in healthy individuals using high density arrays demonstrated that overall median CNV length is less than 2.9 kb, 95% of CNVs are less than 100 kb, and 1%–2% of CNVs in normal individuals are larger than 1 Mb24252627). Another recent study, which reconstructed the genome of 2,504 individuals from 26 populations using a combination of low-coverage whole-genome sequencing, deep exome sequencing, and dense microarray genotyping revealed in total over 88 million variants including 84.7 million single nucleotide polymorphisms, 3.6 million short insertion/deletion, and 60,000 structural variants2829).
Despite higher diagnostic yield, chromosomal microarray testing has not yet been routinely applied as a screening test in patients with developmental delay owing to the perception that it is expensive. However, when compared to the combined cost of a G-banded karyotype, a subtelomeric FISH analysis, and additional other workups, chromosomal microarray testing is not expensive30). The present study also showed that positive yield of genetic tests such as FISH for known genetic disorders and testing for metabolic disorders, was very low (5.9% and 0%, respectively), although positive yield of brain MRI (16%) was similar to that of array CGH. In addition, the abnormal results of neurophysiologic studies did not reveal the etiology of developmental delay despite high positive yield. These results suggest that array CGH is useful in diagnosing patients with developmental delay with respect to cost-effectiveness, because the higher diagnostic yield of array CGH compared to previous conventional diagnostic tests could reduce necessity of additional tests.
The present study has several limitations. First, the number of patients was small. The diagnostic yield of the G-banded karyotyping (11.1%) in this study was significantly higher than those in previous studies (approximately 3%) such that the diagnostic yield of the G-banded karyotyping was not significantly different with those from the array CGH (14.8%). Nonetheless, the results appear to be affected by a sampling bias originating from the small number of the patients in this study, where three patients with chromosomal abnormalities larger than 5 Mb were incidentally included in the study cohort. Second, parental genetic tests for determining whether the CNVs were de novo or inherited could not be performed. Third, diagnostic tests for developmental delay, such as the Bayley Scales of Infant and Toddler Development test, were not performed for each patient, although we included patients with obvious global developmental delay to obtain a higher diagnostic yield from the array CGH. Nevertheless, this study showed the usefulness of applying array CGH in children under 6 years with both unexplained global developmental delay and physical or neurological problems to detect genomic imbalance of less than 5 Mb, which could not be detected in conventional karyotyping. In addition, this study provided summaries of phenotypes from the 5 patients displaying pathogenic and uncertain significant CNVs as detected by array CGH, in conjunction with the locations and sizes of the CNVs. In conclusion, as array CGH can elucidate the cause of developmental delay which was unidentified by past conventional testing, it should also be actively considered as a first-tier diagnostic tool for children with unexplained developmental delay.
Acknowledgments
This work was funded by Ulsan University Hospital (Biomedical Research Center Promotion Fund 11-06).
Notes
Conflicts of interest: No potential conflict of interest relevant to this article was reported.

