Case of mucinous adenocarcinoma of the lung associated with congenital pulmonary airway malformation in a neonate
Article information
Abstract
Congenital pulmonary airway malformation (CPAM), previously known as congenital cystic adenomatoid malformation, is a rare developmental lung abnormality associated with rhabdomyosarcoma, pleuropulmonary blastoma, and mucinous adenocarcinoma of the lung. We report an unusual case of a 10-day-old male newborn with a left lower lobe pulmonary cyst who underwent lobectomy, which revealed type II CPAM complicated by multifocal mucinous adenocarcinoma. KRAS sequencing revealed a somatic mutation in Codon12 (GGT → GAT), suggesting the development of a mucinous adenocarcinoma in the background of mucinous metaplasia. Mucinous adenocarcinoma is the most common lung tumor associated with CPAM, but it generally occurs in older children and adults. Further, all cases in the literature are of type I CPAM. This case in a neonate indicates that malignant transformation can occur very early in type II CPAM.
Introduction
Congenital pulmonary airway malformation (CPAM), formerly known as congenital cystic adenomatoid malformation, is a rare developmental lung abnormality, occurring in 1 per 8,300–35,000 live births, characterized by polycystic lesions in the terminal bronchioles.1) In more than half of diagnosed patients, it is asymptomatic and incidentally discovered through chest radiography in adulthood. Only 25% of patients with CPAM exhibit clinical symptoms at birth.23) In symptomatic cases, normal lung tissue is replaced by polycystic lesions, leading to cyanosis and dyspnea, pneumothorax, hemothorax, and infections. In addition, multiple reports have indicated associations between CPAM and malignant neoplasms, such as rhabdomyosarcoma, pleuropulmonary blastoma, bronchioloalveolar carcinoma, and mucinous adenocarcinoma.4)
Stocker et al.5) categorized CPAM into 5 subtypes according to gross appearance and histological characteristics. Among the five subtypes, type I is predominant, and has been associated with malignancy.6) The present case is the first report of mucinous adenocarcinoma in a Korean neonate with type II CPAM.
Case report
A male neonate weighing 3,080 g was born at 39+2 weeks gestational age (GA) via normal vaginal delivery, with Apgar scores of 9 at 1 and 5 minutes of age. Prenatal ultrasonography at GA 22+0 weeks confirmed a polycystic lung lesion measuring 48×60×39 mm and occupying the entire left lower lung lobe, accompanied by a mediastinal shift and diaphragm inversion. Confirmation of progressive scalp edema, ascites, and pleural effusion led to a thoracoamniotic shunt operation. Obstetric ultrasonography at GA 23+4 weeks indicated partial improvement of hydrops fetalis, with no change in the cystic left lung lesion.
Continuously low (60%–70%) oxygen saturation levels, even with administration of 100% oxygen at 5 L/m, necessitated endotracheal intubation of the neonate in the delivery room, followed by ventilator support in the neonatal intensive care unit. We performed chest computed tomography at 9 days of age (Fig. 1).
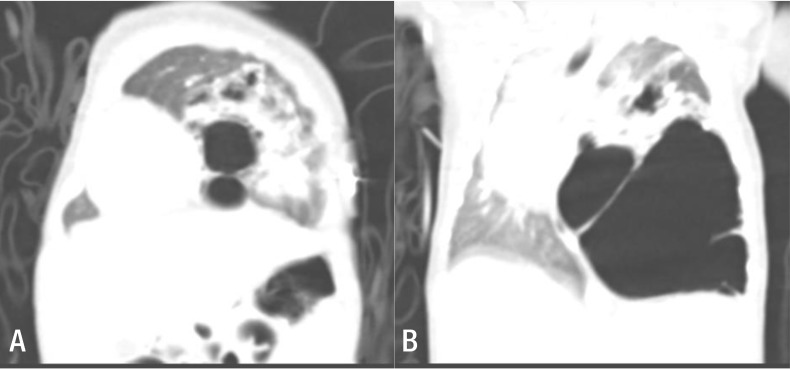
(A) Several large cysts are aggregated on the left lower lobe, with the largest cyst measuring 6 cm in diameter. The left upper lobe has normal features, but is displaced towards the upper section with a decrease in volume due to the mass effect. (B) The mediastinal structures indicate signs of right-sided midline shifting caused by the malformation of the left lower lobe, whereas the right lung has diminished in volume, especially the right upper lobe.
Left lower lobectomy was performed at 10 days of age to remove the aforementioned lesions. At the time of surgery, the left lower lobe was enlarged by the cystic malformation, and the left upper lobe was relatively small. Inspection of the resected lobe revealed polycystic lesions, ranging in diameter from 8 to 20 mm, with the main cyst measuring 35×20 mm. Biopsy revealed normal respiratory epithelial cells coexisting with cells undergoing malignant transformation. These were later confirmed to be mucinous adenocarcinoma (Fig. 2).

(A) Inspection of the resected lobe revealed polycystic lesions, ranging in diameter from 8 to 20 mm, with the main cyst measuring 35×20 mm. (B) Analysis of pathology revealed type II congenital pulmonary airway malformation with extensive mucinous metaplasia. (H&E, ×40)
The KRAS sequence analysis revealed a somatic mutation in codon 12 (GGT → GAT), suggesting development of a mucinous adenocarcinoma in the background of mucinous metaplasia (Fig. 3).
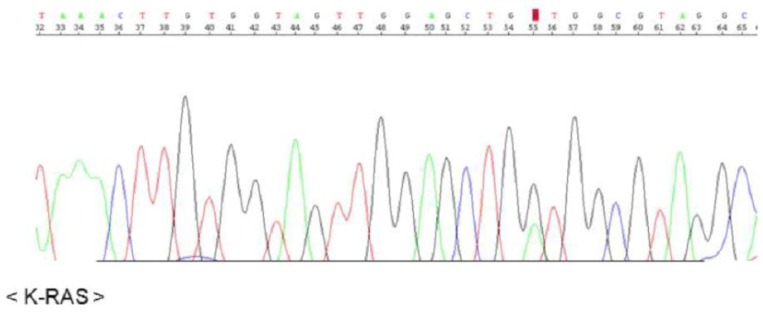
Presence of a mutation in codon 12 of KRAS: G12D (GGT→GAT).
There were no other abnormal findings, aside from the respiratory disorder. The patient was discharged at 32 days postoperatively, and is currently doing well at age 2.4 years.
Discussion
CPAM characteristically presents as a multicystic mass of pulmonary tissue with proliferation of bronchial structures at the expense of alveoli. The specific cause of CPAM is unknown; it is presently considered a hamartomatous malformation or a localized developmental arrest.13) The clinical symptoms of CPAM are diverse. CPAM is asymptomatic in more than half of affected patients and the disease is incidentally discovered on chest radiography, while 25% exhibit clinical symptoms at birth.23) Ruchonnet-Metrailler et al.3) reported that 25% of CPAM patients exhibited respiratory symptoms, with 13% requiring inhaled oxygen, and 11% requiring mechanical ventilation. Symptoms may arise as a consequence of lung immaturity caused by the cystic malformation, with a mediastinal shift, inverted diaphragm, spontaneous pneumothorax, pleural effusion, hemothorax, repetitive mass infections, and hydrops fetalis during the fetal period, due to the pressure effect on the inferior vena cava. Furthermore, associations with malignant neoplasms such as rhabdomyosarcoma, pleuropulmonary blastoma, and adenocarcinoma have been reported. The diagnosis of adenocarcinoma in this pediatric patient was largely decided by KRAS mutation. There was no previously known pathophysiology relating to CPAM or malignant lung tumors, aside from conjecture that the relationship would likely involve abnormal lung tissue development. As exhibited by research on lung cancer morphogenesis in adults, KRAS mutation is associated with epithelial growth factor receptor functions, affecting cell growth, differentiation, and apoptosis. This mutation is especially associated with non-small-cell lung cancers, mainly adenocarcinoma, in adults.7) Corresponding pediatric research has identified cases in which KRAS mutation was found in patients with mucinous adenocarcinoma associated with CPAM.89101112) We therefore concluded that abnormal lung tissue development in neonates due to KRAS mutation is a significant factor associated with CPAM and other malignant lung tumors.
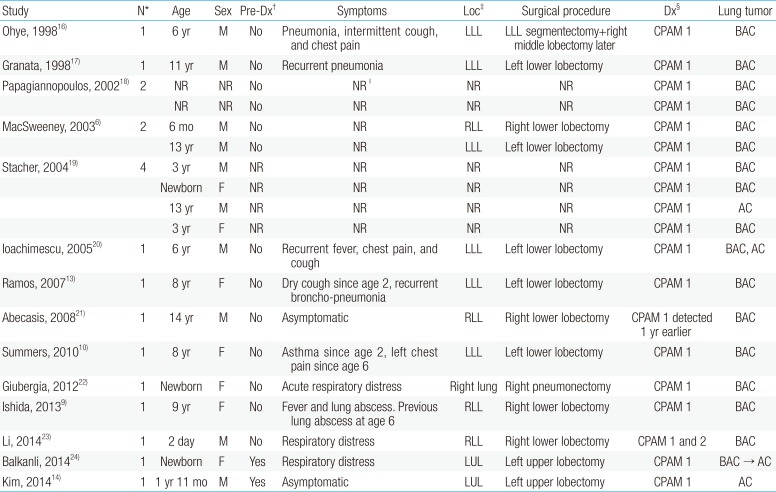
Previous studies indicate that 4%–10% of all lung malignancies that affect children and adolescents are associated with cystic malformations.8) Studies on associated malignant tumors indicate that CPAM is the most prevalent congenital cystic lung malformation, and adenocarcinoma related to type I CPAM is especially predominant. Previous reports on adenocarcinoma by Casagrande and Pederiva,4) Ramos et al.,13) and Kim et al.14) concluded there was an association with type I CPAM (Table 1). Of 39 reported cases of malignant tumors caused by CPAM in patients aged <18 years, 18 were asymptomatic or only exhibited coughing. Furthermore, 10 of 18 children with adenocarcinoma did not exhibit notable symptoms. While the patient described in this report was surgically treated because of his symptoms, asymptomatic pediatric patients with CPAM should undergo regular radiological monitoring, with consideration of elective surgery if necessary.15)

Summarized clinical presentation of cases of MAC arising in CPAM in children reported before 2016
To the best of our knowledge, this is the first case report of mucinous adenocarcinoma, associated with KRAS mutation, in a Korean newborn with type II CPAM diagnosed via lung biopsy. This case may provide the foundation for future research on the potential relationships between CPAM and malignant transformation associated with KRAS mutation.
Notes
Conflict of interest: We have no relevant financial relationships to disclose or conflicts of interest to report.