Concomitant occurrence of Turner syndrome and growth hormone deficiency
Article information
Abstract
Turner syndrome (TS) is a genetic disorder in phenotypic females that has characteristic physical features and presents as partial or complete absence of the second sex chromosome. Growth hormone deficiency (GHD) is a condition caused by insufficient release of growth hormone from the pituitary gland. The concomitant occurrence of TS and GHD is rare and has not yet been reported in Korea. Here we report 2 cases of TS and GHD. In case 1, GHD was initially diagnosed. Karyotyping was performed because of the presence of the typical phenotype and poor response to growth hormone therapy, which revealed 45,X/45,X+mar. The patient showed increased growth velocity after the growth hormone dose was increased. In case 2, a growth hormone provocation test and chromosomal analysis were performed simultaneously because of decreased growth velocity and the typical TS phenotype, which showed GHD and a mosaic karyotype of 45,X/46,XX. The patient showed spontaneous pubertal development. In female patients with short stature, it is important to perform a throughout physical examination and test for hormonal and chromosomal abnormalities because diagnostic accuracy is important for treatment and prognosis.
Introduction
Turner syndrome (TS) is defined as a genetic disorder among phenotypic females that manifests with characteristic physical features and partial or complete absence of the second sex chromosome1). Short stature and hypogonadism are principal signs of TS23). TS affects approximately 1 individual in every 2,500 women14). Nearly half of the individuals with TS have the typical 45,X karyotype while the rest have various types of chromosomal abnormalities including isochromosome and mosaicism125).
TS may largely be diagnosed prenatally or during the neonatal, childhood, or adolescent period3). Cystic hygroma on ultrasonography during pregnancy raises the possibility of TS. Diagnostic screening for TS may be performed with the use of prenatal triple screening tests (human chorionic gonadotropin, alpha fetoprotein, and unconjugated estriol levels). TS may be suspected in cases of clinical manifestations such as hygroma colli during the prenatal period, lymphedema of the extremities, broad chest with widely spaced nipples, webbed neck or cubitus valgus at birth or during infancy, and coarctation of the aorta detected later in childhood2). TS may be suspected in persons with short stature and amenorrhea, as well as repeated abortion in adolescence or thereafter. The diagnosis of TS is confirmed through chromosomal analysis.
Growth hormone deficiency (GHD) is known to occur due to a lack of growth hormone (GH) secretion from the pituitary gland. Clinical manifestations of GHD are short stature, doll-like face, and truncal obesity6). A diagnosis of GHD is made through a comprehensive combination of clinical phenotypes, GH provocation test, and imaging studies7).
Although a differential diagnosis between TS and GHD must be made among female pediatric patients who are admitted to the hospital with the chief complaint of short stature, several cases with concomitant TS and GHD had been reported891011). Nevertheless, the concurrent development of these two disorders is rare and has not yet been reported in Korea. We report two cases of TS that was confirmed in patients with GHD.
Case reports
1. Case 1
A girl aged 8 years 11 months was admitted to our hospital with the chief complaint of short stature. She was diagnosed with GHD at the age of 7 years 6 months and was treated with a daily recombinant human GH (rhGH) regimen.
She was born at 38 gestational weeks via cesarean delivery and had a birth weight of 2.68 kg. She did not have any perinatal problem. Her medical history was unremarkable and did not show any head trauma, seizure, or infections in the central nervous system. No specific family history was found. The paternal and maternal heights were 169 and 168 cm, respectively.
She was admitted to the local hospital with the chief complaint of short stature when she was 7 years 6 months old. At that time, her height was 111.4 cm (–2.30 standard deviation score [SDS]), body weight was 26.0 kg (0.47 SDS), and body mass index (BMI) was 21.0 kg/m2 (1.89 SDS). Bone age was 6.5 years. The peak GH concentrations were 6.05 ng/mL after stimulation with insulin and 6.17 ng/mL after stimulation with glucagon in the GH provocation test implemented at the local hospital. The levels of insulin-like growth factor-I (IGF-I) and IGF-binding protein-3 (IGFBP-3) were 191 ng/mL and 1,957 ng/mL, respectively (reference range, 54.9–206.4 ng/mL for IGF-I and 1,400–6,100 ng/mL for IGFBP-3). Thyroid test results were within normal levels. Brain magnetic resonance imaging (MRI) revealed partial empty sella syndrome (Fig. 1A). She was diagnosed with GHD associated with the partial empty sella syndrome. Hence, she was treated with rhGH, which was administered at a dose of 0.7 unit/kg/wk. Her growth velocity was 5.7 cm/yr after rhGH therapy.

(A) Partially empty sella (arrow) on the brain magnetic resonance image of case 1. (B) Normal brain magnetic resonance image of case 2.
At the time of her visit to our hospital, her height was 119.7 cm (–1.89 SDS), body weight was 36.5 kg (0.67 SDS), and BMI was 22.7 kg/m2 (1.93 SDS). Her bone age was 7.5 years. On physical examination, the characteristic manifestations of TS were observed, such as cubitus valgus, broad chest with widely spaced nipples, webbed neck, and high and narrow palatal arch. No signs of puberty were found. Chromosomal analysis showed 45,X/45,X+mar, which indicated TS with mosaicism. A horseshoe kidney was detected on renal ultrasonography. Echocardiography showed no abnormality.
After the diagnosis of TS, the GH dosage was increased to 1 unit/kg/wk, which is the recommended dosage. Currently, at the age of 9 years 9 months, her height was 126.2 cm (–1.54 SDS), body weight was 33.7 kg (0.26 SDS), and BMI was 21.16 kg/m2 (1.31 SDS). Her annual growth velocity increased to 7.8 cm/yr.
2. Case 2
A girl aged 12 years 4 months was admitted to our hospital with the chief complaint of short stature. She had underwent rhGH therapy for approximately 2 years in other clinic but did not show good treatment response. She showed signs of puberty, such as breast and pubic hair development, at the age of 11 years, although an impaired growth spurt was apparent with the annual growth rate of 3.7 cm/yr.
She was born at 38 gestational weeks via vaginal delivery and had a birth weight of 3.0 kg. She did not have any perinatal problem. She had a history of recurrent otitis media. She did not have any head trauma, seizure, or infections in the central nervous system. No specific family history was found. The paternal and maternal heights were 168 and 162 cm, respectively.
At her first visit to our hospital, her auxological data were as follows: height, 140.1 cm (–1.72 SDS); body weight, 39.7 kg (–0.46 SDS); and BMI, 20.2 kg/m2 (0.48 SDS). Her bone age was 14–15 years. On physical examination, the sexual maturity rates of the breasts and pubic hair were Tanner stages 3 and 2, respectively. In addition, physical findings of webbed neck, low posterior hairline, cubitus valgus, malocclusion, shield-like chest, and fourth metacarpal shortening were observed.
In the GH provocation test, the peak GH concentrations were 7.38 ng/mL after stimulation with L-dopa and 6.26 ng/mL after stimulation with clonidine. The IGF-I and IGFBP-3 levels were 520 ng/mL and 5,670 ng/mL, respectively (reference range, 188.4–509.9 ng/mL for IGF-I and 2,700–8,900 ng/mL for IGFBP-3). The thyroid test results were within the normal levels. The results of the brain MRI, renal ultrasonography, and echocardiography were normal (Fig. 1B). Chromosomal analysis revealed a 45,X/46,XX karyotype.
After the diagnosis of TS with GHD, she underwent rhGH therapy at a dose of 1 unit/kg/wk. Later, she experienced menarche at the age of 12 years 7 months. After 1 year of the rhGH treatment, her growth velocity was 3.8 cm/yr.
Discussion
Short stature is defined as height that is 2 standard deviations below the mean height for age and sex. The treatment, therapeutic effects, and prognosis may be different depending on the etiologies of the short stature. Thus, an accurate diagnosis is imperative. An accurate diagnosis necessitates meticulous physical examinations and appropriate tests6). TS was confirmed through karyotyping in a female with typical phenotypes and GHD was diagnosed using a GH provocation test in a subject with short stature and other clinical findings36).
The differences between TS and GHD included delayed bone age, the peak GH concentration after GH provocation, hypogonadism, and physical findings. Bone age is consistent with chrnological age in patients with TS. However, bone age is significantly lags behind chronological age in patients with GHD. In our cases, bone age was delayed in case 1, whereas bone age was advanced in case 2. In case 2, bone age advancement was mainly owing to spontaneous pubertal development. Up to 30% of TS patients are known to undergo spontaneous pubertal development12).
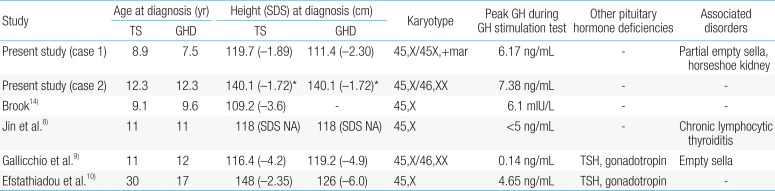
TS could accompany endocrine disorders, including diabetes mellitus and autoimmune thyroid disease. TS patients are known to be GH sufficient. Twenty-four-hour GH secretory profiles and IGF-I levels in TS patients were similar to those in normal children13). The causal relation between of TS and GHD has not been fully understood. The cause of short stature in TS is haploinsuffiency of short-stature homeobox-containing gene in chromosome X. A GH provocation test should be performed in patients whose growth pattern is abnormal compared to the growth reference for TS. Concomitant GHD has been reported in several TS cases. To our knowledge, the first reported case of TS with GHD was in a 9.1-year-old girl, by Brook in 197814). The girl's height was 109.2 cm (−3.6 SDS), and growth velocity was 2.5 cm/yr (Table 1). The peak GH concentration in a provocation test was 6.1 mIU/L after hypoglycemia. Efstathiadou and Tsatsoulis10) reported a 30-year-old woman with TS who was previously diagnosed with multiple pituitary hormone deficiency. Gallicchio et al.9) reported a case of TS with the 45,X/46,XX karyotype and multiple pituitary hormone deficiency. Jin et al.8) reported an 11-year-old girl with TS, GHD, and hypothyroidism.

Summary of cases of concurrent Turner syndrome (TS) and growth hormone deficiency (GHD)
In our study, case 1 was initially diagnosed as GHD. Karyotyping was performed due to the typical phenotype and poor response to rhGH therapy, which revealed 45,X/45,X+mar. In case 2, the GH provocation test and karyotyping were performed simultaneously because of decreased growth velocity and TS phenotype. In the case reported by Brook14), the patient was diagnosed with TS and then GHD according to the decreased growth velocity. The case reported by Efstathiadou and Tsatsoulis10) was confirmed as TS 13 years after the GHD diagnosis. The case reported by Jin et al.8) was diagnosed simultaneously as TS and multiple pituitary hormone deficiency.
In TS patients, rhGH therapy is indicated to attain normal adult height. The recommended rhGH dose is higher in TS than in GHD. Therefore, growth response could be poor in patients with TS who had been initially diagnosed with GHD. In prepubertal TS patients growth velocity was 4.7±1.9 cm/yr before rhGH therapy and 7.6±1.9 cm after 1 year of administration with a rhGH dose of 0.3 mg/kg/wk15). In this study, growth velocity increased from 5.7 to 7.8 cm/yr after the rhGH therapy in case 1. However, our second case did not show a significant increase in growth velocity from 3.7 to 3.8 cm/yr because of the advanced bone age. Hence, early diagnosis is important.
In conclusion, to our knowledge this is the first report of TS and GHD in Korea. TS and GHD are important differential diagnoses in girls with short stature. Though a TS or GHD diagnosis may have been made, a throughout physical examination and further diagnostic tests are important when concomitant TS and GHD are clinically suspected.
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.