Megalencephaly-capillary malformation-polymicrogyria syndrome: the first case report in Korea
Article information
Abstract
Megalencephaly-capillary malformation-polymicrogyria syndrome (MCAP), previously known as macrocephaly-cutis marmorata telangiectatica congenita and macrocephaly-capillary malformation syndrome, is a rare multiple-malformation syndrome that is characterized by progressive megalencephaly, capillary malformations of the midline face and body, or distal limb anomalies such as syndactyly. Herein, we report a female infant case that satisfies the recently proposed criteria of MCAP and describe the distinctive neuroradiological and morphological features. We have also reviewed recently published reports and the diagnostic criteria proposed by various authors in order to facilitate the clinical diagnosis of these children in pediatric neurology clinics.
Introduction
Megalencephaly-capillary malformation-polymicrogyria syndrome (MCAP) is a rare genetic disorder characterized by macrocephaly, capillary malformation, and developmental delay. In 1997, MCAP was first described as macrocephaly-cutis marmorata telangiectatica congenita (M-CMTC) by Clayton-Smith et al.1) and Moore et al.2). Then in 2007, Toriello and Mulliken3) and Conway et al.4) renamed M-CMTC as macrocephaly-capillary malformation syndrome (MCM).
Many diagnostic criteria have been proposed by several authors56). Recently, Mirzaa et al.7) suggested the use of the term MCAP rather than MCM to reflect the large brain size, rather than simply the head circumference and to emphasize the frequency and importance of perisylvian polymicrogyria. Recent diagnostic advances in genetic techniques began to reveal the causative genes associated with the PI3K-AKT pathway in these patients8910). In our study we report the first reported Korean patient who satisfies the new criteria proposed by Mirzaa et al.7) We also reviewed the previous clinical criteria and recent genetic advances regarding this syndrome.
Case report
A 1 year and 7-month-old female had been born to a Korean couple at 39 weeks of gestational age via cesarean section and had a birth weight of 3,540 g. Her perinatal course was uneventful. She visited our outpatient clinic due to developmental delay and an abnormal gait. There was no family history of neurologic disease or developmental delay, and her older male sibling also showed normal development.
On physical examination, she was found to be macrocephalic with 51.2 cm of head circumference (97th percentile), 12 kg of body weight (80th percentile), and 80.8 cm of height (30th percentile). She had an open anterior fontanelle and right-side dominant facial asymmetry (Fig. 1A) as well as mild truncal asymmetry with right-sided hypertrophy. There was neither syndactyly nor polydactyly. Multiple telangiectasia on the skin were found on her nose and upper extremities and hypopigmented, linear skin lesions were found on all of her extremities (Fig. 1B). However, there were no focal neurologic abnormalities, and her gait was relatively stable. She had hyperextensible joints (hip abduction up to 180 degrees) and showed slightly decreased muscle tone (Fig. 1C).
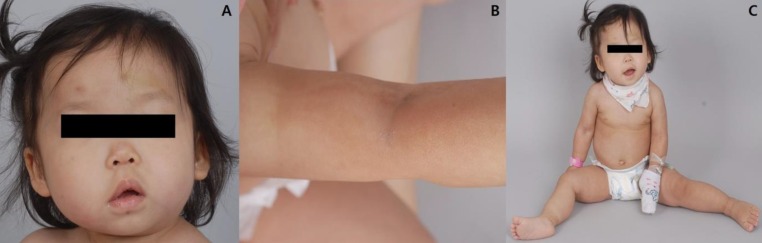
Physical features of the patient. (A) Right-sided facial hypertrophy with multiple brownish patches on the face. (B) Multiple hypopigmented linear lesions on an upper extremity. (C) Hyperextensible hip joint.
She showed language developmental delay (language score, developmental quotient [DQ]=57.9) on the Korean Infant and Child Developmental Test. Her ophthalmologic examination was normal. Even though no clinical seizure was reported, electroencephalography indicated frequent, sharp wave discharges from the left or right frontal areas and a few episodes of diffuse spike and slow wave bursts (Fig. 2). Cerebral magnetic resonance imaging showed Chiari malformation with foraminal stenosis and upper cervical cord compression (Fig. 3A), polymicrogyria in the left frontoparietal lobe (Fig. 3B), and developmental venous anomaly with prominent venous structures in both cerebral convexities (Fig. 3C).

Electroencephalography showed frequent sharp wave or spike discharges from the left or right frontal areas, sometimes associated with exaggerated sleep spindles.
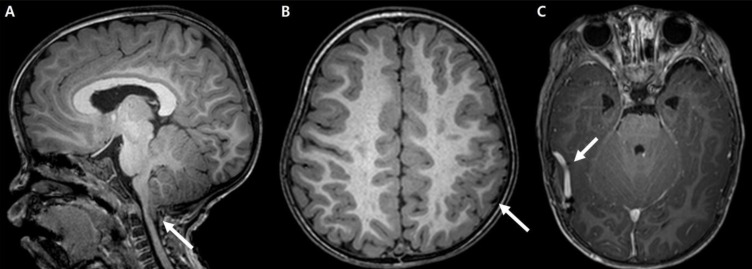
Cerebral magnetic resonance imaging findings for the patient. (A) Chiari I malformation with upper cervical cord compression (arrow). (B) Polymicrogyria (arrow) in the left frontoparietal lobe. (C) Prominent venous structures (arrow) on both cerebral convexities.
Based on the clinical and neuroradiological findings, we diagnosed MCAP (Table 1) and provided rehabilitation therapy for her developmental delay. Regarding Chiari malformation, we planned to wait and see regarding consultation with a pediatric neurosurgeon. On recent follow-up at the age of 5 years and 8 months, her head circumference measured 57 cm which is still greater than the 97th percentile. Despite active language and occupational therapy, she showed mild global developmental delay with moderate language developmental delay (language score DQ=53).

Diagnostic criteria for MCM and MCAP
Discussion
MCAP is characterized by a spectrum of anomalies including primary megalencephaly, prenatal overgrowth, brain and body asymmetry, cutaneous vascular malformations, digital anomalies, connective tissue dysplasia involving the skin, subcutaneous tissue and joints, and cortical brain malformations such as polymicrogyria7). It is a rare genetic disorder and only 130 cases have been reported since it was first introduced as M-CMTC by Clayton-Smith et al.1) and Moore et al.2) in 1997. They proposed that M-CMTC is a distinct disorder exhibiting cutis marmorata, nevus flammeus, cavernous hemangiomas, an asymmetric growth pattern, central nervous system malformations, and neurologic abnormalities.
Since then many cases of M-CMTC have been reported, and the associated anomalies, such as cortical dysplasia, Chiari malformation, and ventriculomegaly were proposed111213). A longitudinal analysis performed in 2007 reported that ventriculomegaly, cavum septum pellucidum or cavum vergae, cerebellar tonsillar herniation, cerebral and/or cerebellar asymmetry, thickened corpus callosum, cortical dysplasia, and polymicrogyria are the main neuroimaging findings of M-CMCT4). During the same year, M-CMTC was renamed MCM as the skin lesions were neither cutis marmorata nor CMTC, but a type of capillary malformation which never improves or ulcerates and is sometimes associated with hypertrophic changes3).
The diagnostic criteria for this syndrome have been proposed by many clinical groups, and Table 1 shows the commonly used diagnostic criteria567). In their proposal of the diagnostic criteria, Mirzaa et al.7) suggested renaming MCM as MCAP. This new term reflects the very large brain size, and which replaces the former term that simply indicated a large head size and also highlights the importance of the polymicrogyria frequently found on neuroimaging7).
Riviere et al.9) recently reported that the de novo germline or postzygotic mutations in the AKT3, PIK3R2, PIK3CA genes are related to MCAP, and thus suggesting the critical role of PI3K/AKT signaling in vascular, limb, and brain development. They also reported that the familial cases of MCAP suggest the autosomal recessive inheritance or germline mosaicism in parents9). De novo CCND2 (encoding cyclin D2) mutations8) and germline activating AKT3 mutations10) are also found in patients with MCAP, and which confirms the PI3K-AKT-related mechanisms in the development of megalencephaly syndrome.
PI3Ks and AKT kinases are important signaling molecules within the PI3K-AKT pathway. They regulate cell growth, proliferation, survival, migration, metabolism, angiogenesis, apoptosis, tumorigenesis, and, in particular, has an important role in brain development, synaptic plasticity, and neurodevelopment891014). As AKT kinase which is essential for cell growth, glucose homeostasis, and the number and size of brain cells is associated with MCAP, characteristics of MCAP might not be limited tomorphological features, but may also have endocrinological manifestations such as glucose instability, and a previously published report supports this relationship patients with MCAP caused by a germline AKT3 mutation10). This new understanding of the signaling pathway in neurodevelopment and overgrowth syndromes will guide the accurate genetic diagnosis and help with the development of ultimately beneficial and successful treatment strategies.
Even though we could not confirm the gene mutation in our patient, we introduced the first case of MCAP in Korea with typical clinical and radiological features and recent diagnostic criteria in other literature studies were also reviewed. Based on the current diagnostic criteria, careful examination and evaluation should be done when encountering patients with suspected MCAP. This careful clinical delineation can then further reveal the heterogenous and common genetic basis of this disease.
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.