Long-term clinical course of a patient with mucopolysaccharidosis type IIIB
Article information
Abstract
Mucopolysaccharidosis type III (MPS III) is a rare genetic disorder caused by lysosomal storage of heparan sulfate. MPS IIIB results from a deficiency in the enzyme alpha-N-acetyl-D-glucosaminidase (NAGLU). Affected patients begin showing behavioral changes, progressive profound mental retardation, and severe disability from the age of 2 to 6 years. We report a patient with MPS IIIB with a long-term follow-up duration. He showed normal development until 3 years. Subsequently, he presented behavioral changes, sleep disturbance, and progressive motor dysfunction. He had been hospitalized owing to recurrent pneumonia and epilepsy with severe cognitive dysfunction. The patient had compound heterozygous c.1444C>T (p.R482W) and c.1675G>T (p.D559Y) variants of NAGLU. Considering that individuals with MPS IIIB have less prominent facial features and skeletal changes, evaluation of long-term clinical course is important for diagnosis. Although no effective therapies for MPS IIIB have been developed yet, early and accurate diagnosis can provide important information for family planning in families at risk of the disorder.
Introduction
Mucopolysaccharidosis type III (MPS III), also known as Sanfilippo syndrome, is a rare autosomal recessive disorder caused by impaired lysosomal degradation and subsequent lysosomal storage of heparan sulfate1). Depending on the deficient enzyme, 4 subtypes (A, B, C, D) are distinguished. MPS III predominantly affects the central nervous system and is characterized by progressive mental deterioration and behavioral problems, whereas individuals with MPS III have less prominent facial features and skeletal changes than those with other types of MPS patients. Therefore, this often leads to a considerable delay in diagnosis. The incidence of MPS III is variable between countries12). Although overall prevalence of patients with MPS in Korea remains elusive, a single center study of 80 patients with MPS showed that Hunter syndrome is the most prevalent (64%) and followed by MPS III (18%)3).
MPS IIIB (OMIM #252920) results from a deficiency of the enzyme alpha-N-acetyl-D-glucosaminidase. The NAGLU gene encoding alpha-N-acetyl-D-glucosaminidase was identified in 19964). To date, more than 120 different disease-causing variants have been reported (www.hgmd.org). Since the 4 types of MPS III remain difficult to distinguish by clinical phenotype, diagnosis should be made by enzyme activity assay. There have been reported some cases of patients with MPS IIIB in Korea35). However, because of the rarity of MPS IIIB, there are few reports on the long-term course of MPS IIIB. Here, we reported a case of a patient with MPS IIIB with presented with neurodevelopmental deterioration and followed up for 20 years. Thus, this report provides a long-term clinical course of MPS IIIB.
Case report
A 4-year-old male was referred to a genetic center due to speech delay. He was born at full-term with a birth weight of 2,900 g, to healthy nonconsanguineous Korean parents. The family including his younger brother had no history of metabolic disorders or neurologic disease. Newborn screening test was normal. He showed normal development until 3 years of age. He was able to walk alone and climb stairs, but he could speak only few words. On examination, he showed mild facial coarseness and hypertrichosis (Fig. 1A). Hepatosplenomegaly was noted. His weight, height, and head circumference were 18.1 kg (-0.67 standard deviation score [SDS]), 96 cm (-1.69 SDS), and 52 cm (0.91 SDS), respectively. Opthalmologic and otolaryngologic examinations revealed no abnormalities. A skeletal survey showed dysostosis multiplex including anterior inferior beaking of lumbar vertebrae and bullet appearance of proximal metacarpal bone (Fig. 2). Screening test for MPS was carried out by qualitative test of urinary glycosaminoglycans by cetyltrimethylammonium bromide and the test result was positive. Due to enzyme replacement therapy (ERT) was available for MPS I, we initially performed α-L-iduronidase assay and result was normal. He was lost to follow-up without specific diagnosis.
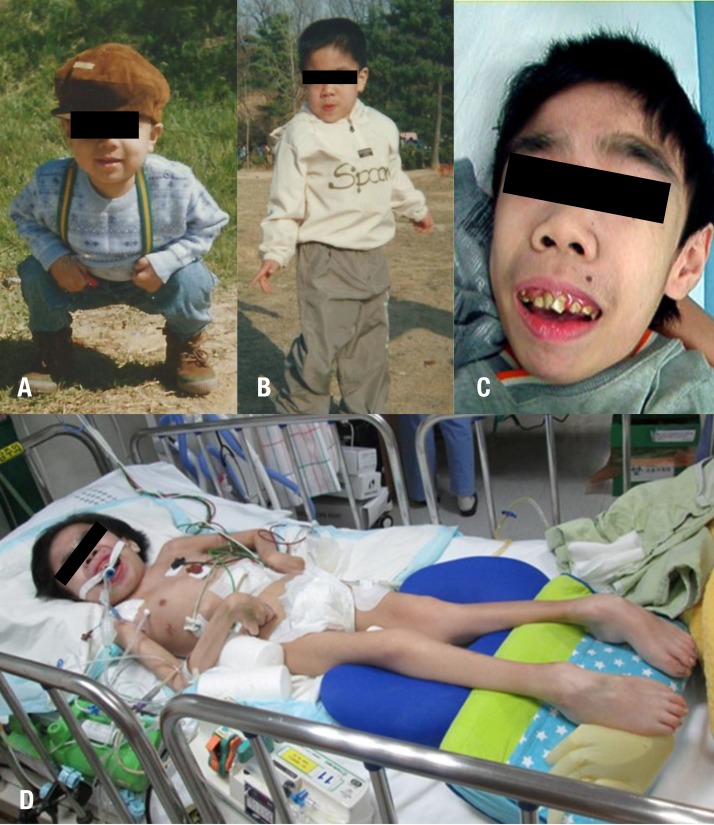
Serial photographs of the patient. (A) Mild facial coarseness was noted at the age of 4 years. Facial dysmorphism, including coarse facial features, a depressed nasal bridge, prominent eyebrows, and malocclusion, was apparent at ages 13 (B) and 18 years (C). (D) He was bedridden and malnourished at the age of 21 years.
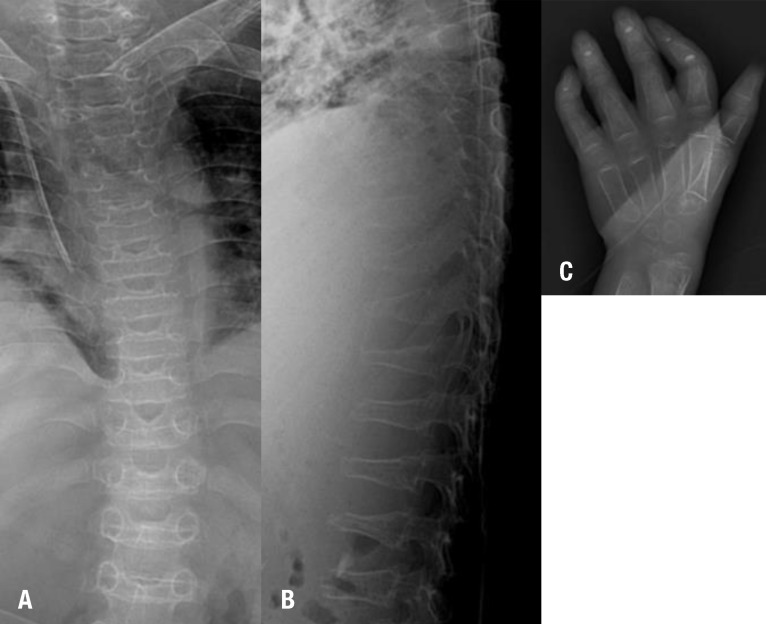
Radiographic image of the dysostosis multiplex. Spine radiographic images of the platyspondyly (A) and anterior beaking (B). (C) A hand radiographic image of the bullet appearance of the proximal metacarpal bone.
He revisited the center with aspiration pneumonia and motor dysfunction at 13 years of age (Fig. 1B). He exhibited behavioral problems such as aggressiveness, anxiety, and sleep disturbance throughout childhood. On opthalmologic examination, corneal clouding was absent at that time. The clinical symptomology including behavioral change, and progressive mental and motor deterioration had raised the suspicion of MPS III. Since then, motor and mental functions had progressively declined, thus he could not sit alone or walk. In addition, communication function has been lost. Epilepsy developed at 18 years of age (Fig. 1C) and he has been hospitalized due to recurrent epilepsy and pneumonia. We determined to make a confirmative diagnosis at that time. Thin layer chromatography revealed excessive excretion of heparin sulfate and NAGLU enzymatic activities were significantly decreased in leukocyte (not detected, range, 0.90–1.51 nmol/hr/mg protein) and plasma (0.14, reference rage, 22.3–60.9 nmol/hr/mL protein), respectively. Direct NAGLU sequencing identified compound heterozygous c.1444C>T (p.R482W) and c.1675G>T (p.D559Y) variants (Fig. 3). The p.R482W mutation was previously reported6). The p.D559Y variant was novel and predicted to be significantly damaging by in silico analysis using SIFT (http://sift.jcvi.org) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/). His parents and younger brother are heterozygous carrier (Fig. 3). He showed spastic quadriplegia with severe cognitive dysfunction and was assumed at the end stage of the disease (Fig. 1D) and is currently 21 years of age.
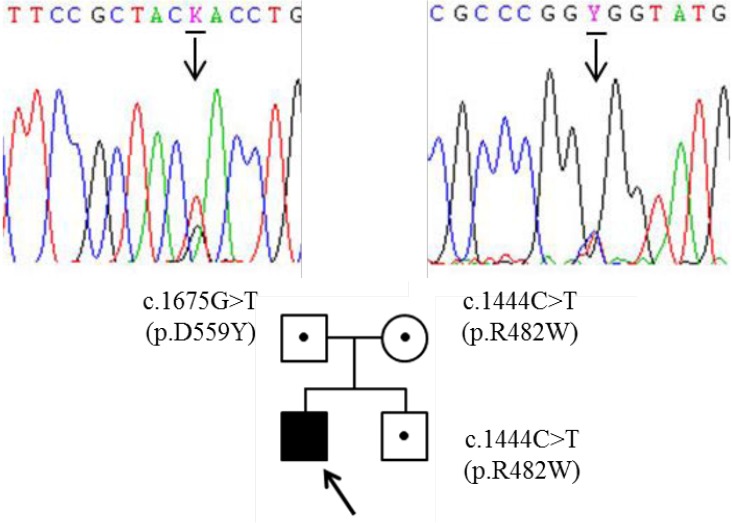
Partial DNA sequences of the NAGLU gene. The patient was a carrier of the compound heterozygous for c.1444C>T (p.R482W) and c.1675G>T (p.D559Y) in NAGLU. Both of his parents and his younger brother were also heterozygote carriers.
The study was granted exempt status by The Institutional Review Board of Samsung Changwon Hospital (IRB study #2014-SCMC-044-00).
Discussion
MPS III has a unique clinical course. Early development is typically normal or near normal, but developmental regression or behavioral problem present between the ages of 2 to 6 years1). In addition, sleep disturbances are frequent among the patients. Therefore, patients in early childhood have been misdiagnosed with idiopathic developmental delay, attention deficit/hyperactivity disorder, or autism spectrum disorders7). Progressive mental deterioration and loss of motor function usually occur after the first decade of life, but degree of mental regression varies among the patients189). The main long-term consequences of MPS III are loss of initiative and progressive motor retardation and subsequent early death in second or third decade of life1). Our case emphasizes MPS III should be considered in the differential diagnosis in patients with behavior problem and developmental regression particularly with facial dysmorphism or skeletal abnormalities. In addition, long-term follow-up as well as clinical suspicion is necessary for diagnosis.
The estimated incidence is very low (0.3–1.9 in every 100,000 live birth) and has a large variation among the countries1210). Heron et al.10) determined that estimated annual numbers of MPS IIIB was between 0.1–2.4 in some European countries, Australia, and Taiwan by organization of previous published studies. MPS IIIA is more prevalent in some European countries including France, Germany, Netherlands, and Sweden and Australia, but MPS IIIB is more prevalent in Portugal and Taiwan2101112). Also, the incidence of MPS IIIB is unknown in Korea. Eight patients with MPS IIIB have been reported in Korea, and of them, enzymatic assay and molecular study were performed in only one patient35). However, this disease may underestimate owing to the phenotypic heterogeneity and relatively mild somatic features. Further nationwide survey of MPS is needed to verify the estimated incidences in Korea.
The NAGLU gene consists of six exons and encodes a 720 amino acid protein4). To date, more than 120 different disease-causing variants have been reported and the missense, small deletions, and insertions are common mutation types (www.hgmd.org). The R482W previously reported may have a founder effect in the Turkish populations69). Although the functional consequences of the mutation were not analyzed using an in vitro assay, D559Y is a highly conserved residue across various species by multiple sequence alignment using ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/). In addition, this variant is not found on the dbSNP (http://ncbi.nlm.nih.gov/SNP/), 1000 Genomes Database (http://browser.1000genomes.org), or ClinVar (http://ncbi.nlm.nih.gov/clinvar/).
With the advent of molecular biology, ERT is available for the treatment of lysosomal storage disease including MPS I, II, and VI13). In addition, MPS IVA has been on clinical trial for ERT14). Since enzyme replacement does not effective neurological symptoms due to the blood-brain barrier, intrathecal enzyme administration has been on clinical trial13). In this respect, early and accurate diagnosis of MPS is imperative to optimize clinical outcomes, particularly for those disorders that are ameliorated by ERT or hematopoietic stem cell transplantation.
In summary, we described progressive clinical course of MPS IIIB with aging. In this case, typical long-term consequence of gradual progressive neurodevelopmental decline or regression with mild somatic features would help to identify more cases with MPS IIIB. Although there are no effective therapies yet in MPS III, early diagnosis is crucial for genetic counseling in at-risk families.
Acknowledgments
The authors wish to thank the patient and his parents.
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.