The first Korean case with Floating-Harbor syndrome with a novel SRCAP mutation diagnosed by targeted exome sequencing
Article information

Abstract
Floating-Harbor syndrome is a rare autosomal dominant genetic disorder associated with SRCAP mutation. To date, approximately 50 cases of Floating-Harbor syndrome have been reported, but none have been reported in Korea yet. Floating-Harbor syndrome is characterized by delayed bony maturation, unique facial features, and language impairment. Here, we present a 6-year-old boy with a triangular face, deep-set protruding eyes, low-set ears, wide nose with narrow nasal bridge, short philtrum, long thin lips, clinodactyly, and developmental delay that was transferred to our pediatric clinic for genetic evaluation. He showed progressive delay in the area of language and cognition-adaption as he grew. He had previously undergone chromosomal analysis at another hospital due to his language delay, but his karyotype was normal. We performed targeted exome sequencing, considering several syndromes with similar phenotypes. Library preparation was performed with the TruSight One sequencing panel, which enriches the sample for about 4,800 genes of clinical relevance. Massively parallel sequencing was conducted with NextSeq. An identified variant was confirmed by Sanger sequencing of the patient and his parents. Finally, the patient was confirmed as the first Korean case of Floating-Harbor syndrome with a novel SRCAP (Snf2 related CREBBP activator protein) mutation (c.7732dupT, p.Ser2578Phefs*6), resulting in early termination of the protein; it was not found in either of his healthy parents or a control population. To our knowledge, this is the first study to describe a boy with Floating-Harbor syndrome with a novel SRCAP mutation diagnosed by targeted exome sequencing in Korea.
Introduction
Floating-Harbor syndrome (OMIM #136140) is an autosomal dominant genetic disorder characterized by short stature, delayed bony maturation, unique facial morphology, and expressive language impairment [1]. Recently, truncating mutations in SRCAP (Snf2 related CREBBP activator protein) have been shown to cause Floating-Harbor syndrome [2]. Till date, approximately 50 cases of SRCAP exon 34 mutations, and two cases in SRCAP exon 33 mutations have been reported [3,4]. Genetic confirmation of Floating-Harbor syndrome has not previously been reported in Korea. This may be because routine genetic testing for SRCAP mutations is not available in most Korean hospitals due to the rarity of this disease. Exome sequencing can be useful in determining causative genetic variants associated with certain diseases, especially the inherited/congenital Mendelian diseases [5]. Here, we present the first Korean child confirmed to have Floating-Harbor syndrome with a novel SRCAP mutation (exon 33), as determined by targeted exome sequencing.
Case report
A 6-year-old boy presenting with rhinorrhea for several months visited our medical center otolaryngologist, who confirmed by nasal endoscopy that he had no anatomical anomaly and diagnosed him with adenoid hypertrophy and chronic sinusitis. The otolaryngologist doubted a congenital problem like Crouzon syndrome because he exhibited a dysmorphic face and developmental delay. Thus, he was transferred to our pediatric clinic for genetic evaluation. He had a triangular face, deep-set protruding eyes, low-set ears, a wide nose with a narrow nasal bridge, short philtrum, long thin lips, and clinodactyly. He was born as the first baby in a set of twins at a gestational age of 34 weeks. At birth, his body weight was 2,030 g (50th–75th percentile), height was 44 cm (25th–50th percentile), and head circumference was 32 cm (50th–75th percentile). The body weight of his twin sister was 2,110 g (50th–75th percentile), height was 47 cm (75th–90th percentile), and head circumference was 32 cm (50th–75th percentile) at birth. He scored as normal on the audiometry test at birth. His parents were healthy without any diseases. As his parents felt that he showed language and growth delays compared to his twin sister’s progress, he had visited another hospital for evaluation when around 1 year old; however, he was not diagnosed with any syndrome, and his karyotype was normal.
At the age of 16 months, he was diagnosed with a mild delay of gross motor and language skills based on the Korean version of Denver Developmental Screening Test (DDST), scoring at 12 months in both categories; at 12 months for gross motor and language, at 16 months for fine motor, personal sociability, and cognition-adaptation. At that time, his body weight, height, and head circumference appeared in the normal range of growth. However, his parents were informed that his growth indexes were much lower than those of his twin sister. At the age of 29 months, his developmental delay was considerably aggravated and showed obvious differences: gross motor at 22 months, fine motor at 27 months, personal sociability at 24 months, language at 16 months, and cognition-adaptation at 16 months by Korean DDST. At that time, his parents stated that he had difficulty concentrating on things and sometimes repeated certain words, suggesting echolalia or a phonological disorder. They also stated that he preferred things to people.
His bone age was estimated to be 60 months, which was significantly younger than his chronological age of 75 months. At that time, his height was 113 cm (10th–25th percentile) and he was much shorter than his twin sister, as per his parents. His growth indices and z scores, according to the 2017 Korean pediatric growth chart after birth, and his bone age are shown in Table 1. The heights of father and mother were 179 cm and 168 cm, respectively; thus, midparental height (MPH) was 180 cm (MPH z score, 0.976). The patient’s predicted adult height (PAH) was 170 cm (PAH z score, -0.77). The baseline growth hormonal tests were performed when he was 6 years and 10 months; insulin-like growth factor-I 163.04 ng/mL (reference, 58.37–212 ng/mL), insulin-like growth factor-binding protein 3 1,795 ng/mL (reference, 1,219–3,710 ng/mL). And other hormonal test were performed when he was 7 years and 2 months; thyroid-stimulating hormone 4.12 mIU/mL (reference, 0.17–4.05 mIU/mL), free T4 1.23 ng/dL (reference, 0.9–2.6 ng/dL), testosterone 0.04 ng/mL (reference, 0.03–1.0 ng/mL), dehydroepiandrosterone-sulfate 45.63 μg/dL (13–115 μg/dL), luteinizing hormone 0.84 mIU/mL (reference, 0.2–0.3 mIU/mL), and follicle stimulating hormone 0.53 mIU/mL (0.36–3.0 mIU/mL).
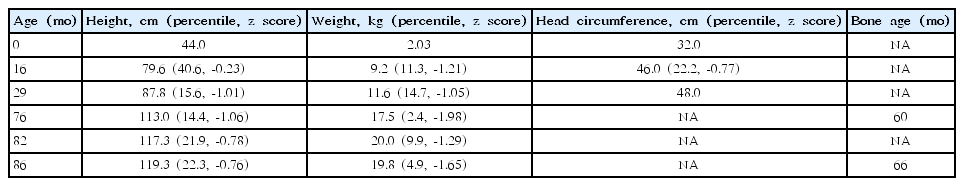
Growth indices according to the 2017 Korean pediatric growth chart, and bone age of the Korean boy with Floating-Harbor syndrome
We conducted language education for him, audiological tests with otolaryngology, along with psychiatric consultation. Speech delay was assessed as a fourth degree of articulation disorder in spoken language evaluation. Hearing tests were performed by auditory brain stem response and otoacoustic emission; results were in the normal range. He had no anatomical anomaly of the brain according to magnetic resonance imaging and no neurological defects at 6 years of age. The ultrasonogram of kidney was normal with no anomaly of the renal system. Ophthalmologic tests are planned for screening.
We conducted targeted exome sequencing for this patient because there are several syndromes and relevant genes related to developmental delays and facial deformities. He initially was suspected to have Crouzon syndrome, but he had some milder morphological signs compared to the classic phenotypes of Crouzon syndrome. For exome sequencing, the written informed consent was obtained from all subjects (patient and his parents) before peripheral blood sampling. This study was approved by the Institutional Review Board of Keimyung University Dongsan Medical Center (approval number: 2017-09-028). A library was prepared with the TruSight One Sequencing Panel (Illumina, San Diego, CA, USA), which enriches samples for approximately 4,800 genes of clinical relevance. Massively parallel sequencing was conducted with NextSeq (Illumina). Common variants with minor allele frequencies ≥1%, according to population databases, were removed to eliminate population-specific polymorphisms.
Finally, a novel SRCAP mutation (c.7732dupT, p.Ser2578Phefs*6 in exon 33) was identified. Validation by Sanger sequencing was also performed (Fig. 1). The mutation was not found in any control population database, nor in his healthy parents (Fig. 1). This is a novel variant and had not been reported previously. In this variant, the protein was terminated early and hence interpreted as a pathogenic variant, since it is neither found in the control population nor in the parents with normal phenotype. Based on the guidelines for interpretation of sequence variants by American College of Medical Genetics and Genomics, and Association for Molecular Pathology [6], this mutation may be considered pathogenic. According to SRCAP mutation and specific phenotypic characteristics, the patient was diagnosed as Floating-Harbor syndrome. Further, pathogenic variant of FGFR1, FGFR2, or FGFR3 associated with Crouzon syndrome was not found in him. Since its diagnosis as an autosomal dominant genetic disease, genetic counseling has been carried out.
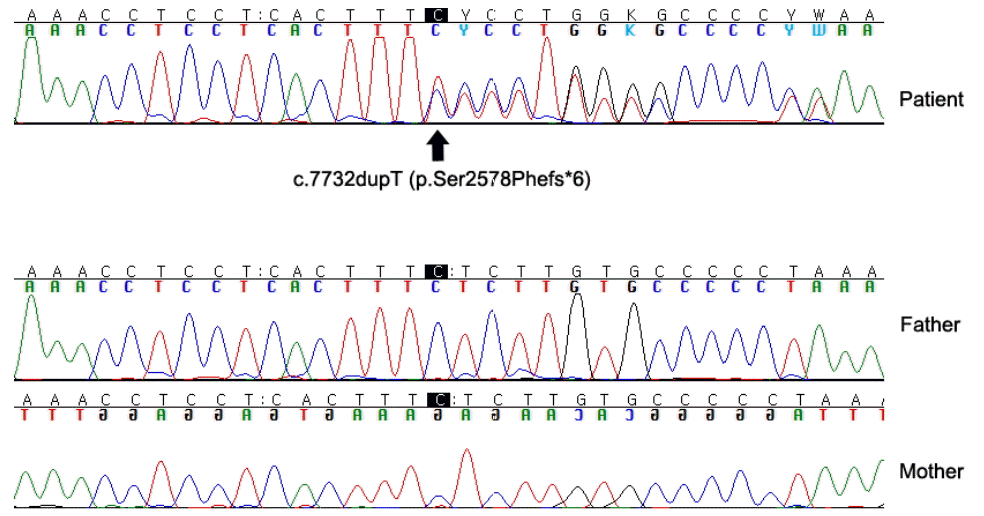
Sanger sequencing validation of the SRCAP mutation (c.7732dupT, p.Ser2578Phefs*6) in exome 33 in a Korean boy with Floating-Harbor syndrome and his parents. The mutation was found neither in his father nor in his mother, both of whom are healthy.
Discussion
Floating-Harbor syndrome is characterized by short stature with delayed bone age and bone mineralization, deficits in expressive language, mental retardation, and skeletal/craniofacial abnormalities [7]. The mean age at diagnosis was 8 years in previous patients [7] while it was 7 years in our patient. The characteristic morphologic features of the Floating-Harbor syndrome include a triangular face, a wide nose tip, short philtrum, deep eyes, long thin lips, and low-set ears [7]. All of the above-mentioned findings were observed in our patient. Among 52 patients with Floating-Harbor syndrome, 25 were found to have delayed bone maturation compared to their chronologic age [7]. Further, 7 of these 25 patients showed compulsive or obsessive behavior while 9 of 32 showed hyperactivity disorder [7]. In our patient, bone age was also delayed accompanied by poor concentration and distracted appearance. Clinical features, such as cleft lip, wide thumb, low birth weight, small head circumstance, cryptorchidism, strabismus, and hearing loss, were reported in some patients with Floating-Harbor syndrome [7], but these features were not observed in our case.
SRCAP, located on chromosome 16p11.2, encodes a SNF2-related chromatin remodeling ATPase that acts as a co-activator for CREBBP [4,8]. CREBBP plays a key role in regulating cell growth, cell division, DNA repair, differentiation, cell death, and tumor suppression [8,9]. The nonsense or frame-shift mutation in exon 34, the last exon of SRCAP, produces a C-terminal-truncated SRCAP protein that has lost its functional domain, leading to a dominant negative effect [4]. The recently reported mutation on exon 33 also produces C-terminal-truncated SRCAP protein similar to that in case of exon 34 [3]. The patients, with mutation in exon 33, most likely exhibit the symptoms of classical Floating-Harbor syndrome, but with relatively milder language delay and cognitive decline to enable normal school life [10]. However, the number of patients with Floating-Harbor syndrome due to exon 33 mutation is still small, and hence differences between them are difficult to observe.
Rubinstein-Taybi syndrome is known to be associated with a mutation in CREBBP [9]. Thus, Floating-Harbor syndrome and Rubinstein-Taybi syndrome are clinically similar and known to be the first to be differentially diagnosed. Rubinstein-Taybi syndrome is characterized by a small head circumstance, protruding forehead, wide-set eyes, low-set ears, thick eyebrows, a wide nose, and a small jaw [11]. These patients also exhibit skeletal anomaly of fingers and delayed bone maturation, strabismus, ptosis, obstruction of nasal passages, and dense teeth [11]. In addition, Rubinstein-Taybi syndrome is characterized by poor growth after birth, short stature, developmental delay, and mental retardation, accompanied by speech disturbance, decreased muscle tension, unstable gait, and epilepsy [11]. Further, Rubinstein-Taybi syndrome may show distracted and repetitive behavior, depression, obsessive compulsive disorder, and autism [11]. Clinical similarity was observed in our patient, including small jaw, wide-set eyes, low-set ears, delayed bone development, language disability, overall developmental delay, and distracted and impulsive nature.
There have been some studies of growth hormone therapy in Floating-Harbor syndrome patients, showing positive effects of growth hormone on the short stature of patients [7,10]. Some patients have achieved heights in the normal range after growth hormone therapy [12]. Other endocrinological abnormalities, such as delayed puberty and precocious puberty, have been reported in few studies, all in females [10,13]. In the case of the patient reported here, his height was within the normal range, though much lesser than that of his female twin. And the PAH is much shorter than the MPH. In summary, we report a case of Floating-Harbor syndrome in a patient with a facial dysmorphism and developmental delay. This is the first report of Floating-Harbor syndrome diagnosed by targeted exome sequencing in Korea.
Notes
No potential conflict of interest relevant to this article was reported.