Application of CRISPR-Cas9 gene editing for congenital heart disease
Article information

Abstract
Clustered regularly interspaced short palindromic repeats and CRISPR-associated protein 9 (CRISPR-Cas9) is an ancient prokaryotic defense system that precisely cuts foreign genomic DNA under the control of a small number of guide RNAs. The CRISPR-Cas9 system facilitates efficient double-stranded DNA cleavage that has been recently adopted for genome editing to create or correct inherited genetic mutations causing disease. Congenital heart disease (CHD) is generally caused by genetic mutations such as base substitutions, deletions, and insertions, which result in diverse developmental defects and remains a leading cause of birth defects. Pediatric CHD patients exhibit a spectrum of cardiac abnormalities such as septal defects, valvular defects, and abnormal chamber development. CHD onset occurs during the prenatal period and often results in early lethality during childhood. Because CRISPR-Cas9-based genome editing technology has gained considerable attention for its potential to prevent and treat diseases, we will review the CRISPR-Cas9 system as a genome editing tool and focus on its therapeutic application for CHD.
Key message
Clustered regularly interspaced short palindromic repeats and CRISPR-associated protein 9 (CRISPR-Cas9) system has made a big step in the genome editing which still requires technical developments for the efficient applications in the many fields including congenital heart diseases (CHDs), closely related to the genomic abnormality. In this review, we tried to cover the most updated researches of CRISPR-Cas9 in the CHDs to understand the current technologies, eventually becoming therapeutic bases for the CHD patients.
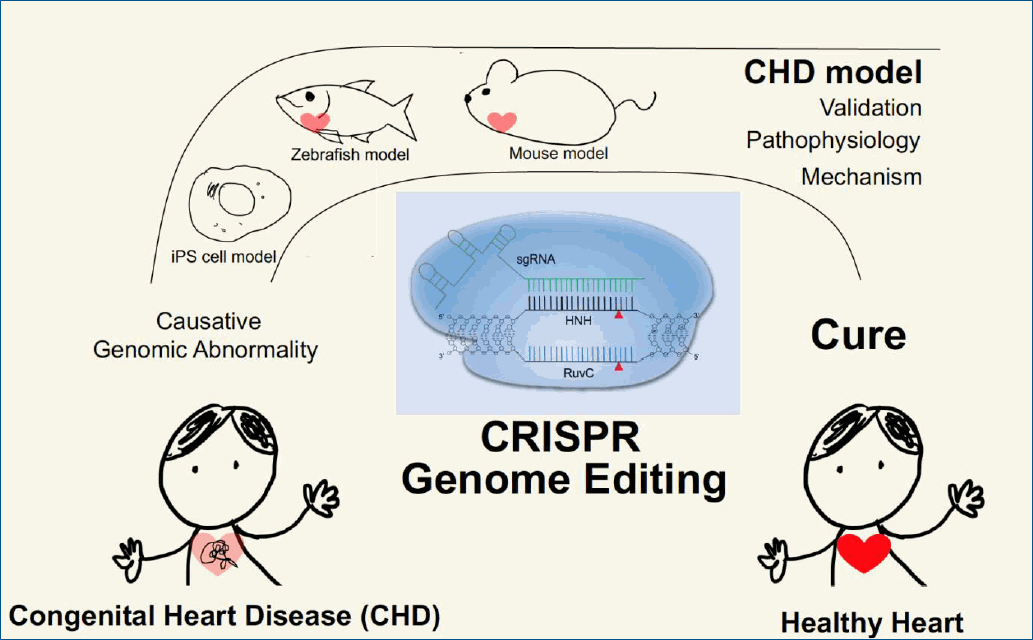
Graphical abstract
Introduction
Congenital heart disease (CHD) is the most commonly diagnosed congenital disorder in newborns—accounting for the highest mortality rate outside of infectious etiologies [1]. Based on a 2017 survey of the regional, national, and global burden of CHD, these diseases account for 261, 247 deaths per year globally [2]. Regionally, the highest annual mortality rate per 100,000 infants (i.e., <1 year old) was assigned to Oceania as estimated by the Cause of Death Ensembl model. Regardless of these national differences, CHD is a critical causative factor of childhood morbidity and mortality globally [3].
Substantial effort has been expended to understand pathogenesis of CHD by investigating genetic variations and molecular pathways. Many of these diseases have now been genetically characterized as a result of technological advancements. For example, aneuploidies (i.e., an abnormal number of chromosomes in cells) [4,5] and microdeletions have been identified using fluorescence in situ hybridization to visualize chromosomal alterations [6-8]. Soon after these studies, single-base mutations were identified using human genomic data. Copy number variants [9] and single nucleotide polymorphisms [10] were subsequently discovered using recently developed high-throughput sequencing technologies [11].
Researchers have pursued countless means to develop, improve, and utilize genome editing strategies to alter genetic mutation-induced diseases, including CHD. These studies began with the discovery of restriction enzymes to create double-strand breaks (DSBs) that allow for genome editing. Several early methods employed zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) to improve the efficiency of editing [12]; however, the discovery of the prokaryotic clustered regularly interspaced short palindromic repeats (CRISPR)-Cas system revolutionized these efforts, as it was more specific, simple, and robust [13]. Consequently, CRISPR-Cas strategies to repair genetic mutations through either nonhomologous end joining or homologous dependent repair with wild-type alleles have been tested for various diseases using model systems [14].
The CRISPR adaptive immune system and the associated RNA-guided Cas nuclease were discovered serendipitously [15-19]. Bacteria possessing less susceptibility to bacteriophage attack were found to have a high homology between a repeating region and the phage genome, which led to the idea that these sequences represented a bacterial defense mechanism [20]. In support of this suggestion, short phage genomic sequences, later called protospacers [21], were found to be incorporated in the bacterial genome cluster [22]. These newly inserted sequences transcribed CRISPR RNAs (crRNAs) [23-25], which enabled Cas-mediated phage genomic cleavage with highly accurate target sequence specificity [25]. In addition to crRNAs, researchers identified additional RNAs necessary for CRISPR-Cas function, called transactivating crRNAs (tracrRNAs) [23], which formed a secondary structure critical for recruiting the Cas protein to the phage genome. In addition to these RNA components, the targeting genome must contain a short recognition site named protospacer adjacent motif (PAM) [26]. which is proximally located and ranges from approximately 2 to 5 nucleotides from the targeting region [27,28]. It was determined that crRNAs and tracrRNAs became functional when they produced a single fused RNA called a single guide RNA (sgRNA) [29]. These components were subsequently reconstituted in Escherichia coli [29] and eukaryotes [30,31].
More than 10 different CRISPR-Cas systems have been identified in bacteria and archaea [13,32,33]. Based on the Cas nuclease types and CRISPR sequences, these systems have been categorized into 2 classes. Class 1 contains type I, III, and IV CRISPR systems, which usually function in a multiple Cas protein complex. Class 2 contains type II, V, and VI CRISPR systems [34,35]. In the latter type, a single DNA endonuclease called Cas9 is responsible for double-stranded DNA cleavage. Among these systems, the most prevalent is the type II CRISPR-Cas9 system, which was discovered in Streptococcus pyogenes. Its popularity results from the simplicity of the Cas9 enzyme (spCas9) and PAM motif (NGG) from this bacterium [36].
In this review, we will focus on the CRISPR-spCas9 system to explain how it works, review applications using disease models, and discuss future research directions aimed at reducing mortality and improving the lives of children affected with CHD.
CRISPR-Cas9 function
The CRISPR-Cas9 system has similarities with RNA interference (RNAi) [33,36]. Both are adaptive immune systems to defend against foreign organisms and both utilize RNA as a homology-based strategy to cleave nucleic acids. RNAi relies on 21 or 22 nucleotide double-stranded RNAs that mediate their activity through a family of endogenous ribonucleoprotein complexes called RNA-induced silencing complexes [37,38] that trigger target RNA degradation [39-41]. Similarly, CRISPR-Cas9 uses small RNAs by combining a crRNA and tracrRNA or using a fused sgRNA to recognize target sequences to produce a double-stranded break. Both RNAi, in the form of small interfering RNAs, and CRISPR-Cas9 are now widely used for both research and therapeutic applications.
The type II locus produces the Cas9 enzyme, which contains 6 functional domains. These domains have a multitude of functions, including binding of guide RNAs and target DNA sequences, interacting with the PAM motif, and cleaving nucleic acids [32,42,43]. The enzyme itself is 1368 amino acids in length and, based on structural studies, the HNH-like nuclease domain and the RuvC domain share similarity with a retroviral integrase superfamily that employs a metal-ion catalytic mechanism that causes a conformational change to control enzymatic activity [32,44]. Based on this information, a conserved histidine (H840) residue in the HNH-like domain and an aspartate (D10) residue (29) in the RuvC domain were established to be critical for cleavage [42,45]. Individual mutations of H840A or D10A cause Cas9 to become a nicking endonuclease (nickase), which have only single-strand cleavage capabilities. Double mutants have no DNA cleavage ability and are referred to as dead Cas9 (dCas9) [29]. Ordinarily, the HNH-like domain cuts the target strand, which is complementary to the sgRNA, and the RuvC domain cuts the opposite, non-target DNA strand [42,46,47]. The C-terminal domain also interacts with the PAM motif in the target genome [26], where 3 base pairs are excised to leave a blunt-ended molecule [29]. The crRNA and tracrRNA binding domains are critical for the exquisite positional genomic selectivity and the subsequent CRISPR-mediated interference.
Transcription of the CRISPR array locus and subsequent enzymatic processing of the precursor RNA (including RNase III-mediated cleavage and 5’ end trimming) result in the generation of a mature crRNA that is approximately 20 nucleotides long. This crRNA contains the foreign nucleic acid recognition segment at the 5’ end and CRISPR repeats at the 3’ end. The latter target the protospacer region next to the PAM (NGG in case of CRISPR-Cas9) [25]. A small noncoding tracrRNA, which is transcribed from the CRISPR locus, can also associate with mature crRNA through CRISPR repeats to form a specific secondary structure that is necessary to recruit Cas9 [23]. The single sgRNA formed by crRNA and tracrRNA fusion has been the focus of efforts to optimize genome editing [29]. A schematic representation of CRISPR-Cas9 cleavage is depicted in Fig. 1.
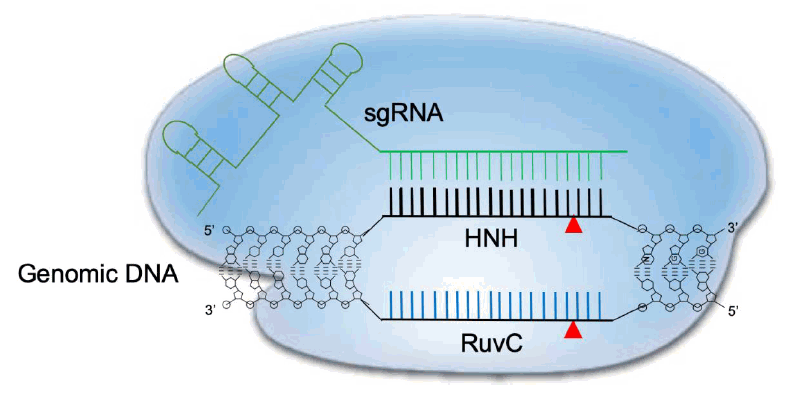
Schematic diagram of CRISPR-Cas9 cleavage of double-stranded DNA. The Cas9 dependent protospacer adjacent motif (PAM) genomic sequence is NGG. A 20-nucleotide long sequence is targeted by a complementary RNA (sgRNA) and structural RNAs responsible for Cas9 enzyme recruitment. Once the sgRNA binds the target sequence, Cas9’s HNH-like endonuclease cuts the 3’ position end of the PAM motif. At the same time, the nontargeting genomic strand is cleaved by the RuvC-like domain in Cas9, leading to a double-strand break. CRISPR-Cas9, clustered regularly interspaced short palindromic repeats and CRISPR-associated protein 9.
Development of genome editing
The CRISPR-Cas9 system is revolutionary because it permits genome editing of specific sequences by insertion, deletion, or substitution using rationally-designed templates [12,48]. Initial attempts to edit the genome used homologous recombination to insert exogenous foreign DNA [49-51]; however, this process was inefficient and poorly controlled; resulting in random and multiple insertional events [51,52]. To find a more position-dependent strategy, researchers used restriction enzymes to target a particular locus to create a DSB. Unfortunately, enzymes that target short sequences (i.e., 4 to 6 nucleotides) [53,54] or long sequences (i.e., 14 to 40 nucleotides) [55], were limited by multiple target recognition sites and repair of DSBs by nonhomologous end joining [56]. As a result, researchers began to focus on engineering nucleases to allow greater positional specificity in editing the genome. A fusion protein using the zinc-finger module paired with the DNA cleavage domain of the Fok1 endonuclease was developed and called ZFNs. These enzymes represented yet another step toward specific editing of DNA sequences [57-59]. The zinc-finger module could bind DNA by sensing a 3 base pair long stretch of DNA, which allowed precise positioning by combining different modules [60-62]. Around the same time, a bacterial transcription activator-like effector (TALE) was being used for a similar purpose. TALE bound DNA by sensing a single nucleotide. When recombined with the Fok1 endonuclease, the engineered TALEN enzyme was created, which also contributed to development of genome editing [63-66]. However, adoption of this technology was limited because every target DNA sequence required de novo molecular engineering of a chimeric protein. Consequently, the CRISPR-Cas9 system solved these issues as it relied on small, noncoding RNAs to exquisitely target specific sequences and it contained strong nuclease activity [48].
Applications of CRISPR-Cas9 for CHD
CHD is generally associated with genomic perturbations combined with environmental effects that often result in an unclear etiology [11]. Although genetic factors play important roles in CHD, detailed information is largely unknown. While, some of these genomic variations are not associated with a phenotype, those that are could benefit from genomic editing to cure CHD patients. The identification of causative genes and mutations in CHDs has accelerated with the recent development of next-generation sequencing technologies. A summary of selected causative CHD mutations in shown in Tables 1 and 2. Table 1 lists CHD phenotypes that have been considered for intervention using CRISPR-Cas9, while Table 2 lists potential mutations associated with CHDs that, if verified, may also benefit from a gene editing approach. Prior to implementation, the development and study of appropriate CHD models are necessary (Fig. 2). Below, we will focus on CRISPR-Cas9 applications using CHD animal models; however, there are many reviews of CRISPR-Cas9 strategies for human heart diseases [14,67-69].

CRISPR-Cas9 in congenital heart diseases (CHDs)

Potential candidates for CRISPR-Cas9 genome editing
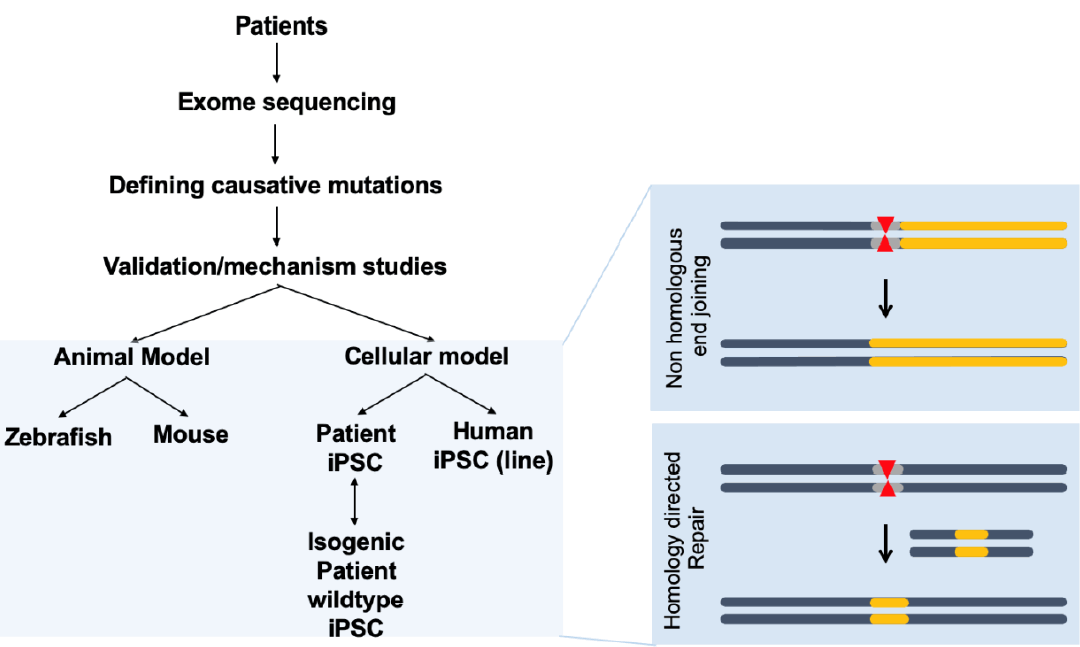
CRISPR-Cas9 applications for human congenital heart disease (CHD). Human samples such as white blood cells can be used for exome sequencing to detect novel genetic variations with single nucleotide resolution. Identification of novel mutations causing CHD can be validated in animal models such as mice or zebrafish, which are amenable to genetic manipulation. Application of CRISPR-Cas editing in both animal models and in vitro systems, such as induced pluripotent stem cells (iPSCs) or differentiated cells derived from iPSCs, can validate the editing design. CRISPR-Cas9, clustered regularly interspaced short palindromic repeats and CRISPR-associated protein 9.
Initially, the development and testing of cardiac-specific CRISPR-Cas9 in vivo focused on mouse models. Carroll and colleagues developed transgenic mice expressing high levels of Cas9 in the heart using the α-myosin heavy chain (Myh6) promoter combined with intraperitoneal injection of adenoassociated virus serotype 9 (AAV9) to deliver an sgRNA targeting Myh6. A single injection of this virus resulted in 75% transduction efficiency causing Myh6 repression [70]. Guo et al. [71] utilized a Cre-LoxP genetic strategy for CRISPR-Cas9 delivery. Using the cardiac troponin T promoter to drive Cre enzyme expression with sgRNAs delivered using AAV9 in neonatal mice, a Cas9-GFP fusion protein incorporated in the Rosa26 locus guaranteed Cre activity-dependent Cas9 expression. With this strategy, cardiac troponin T-dependent gene editing by CRISPR-Cas9 was possible in neonatal mice. In addition, several genes reported to be commonly associated with CHDs (e.g., Myh and serum response factor) were targeted to develop an animal model and to study the pathogenesis of disease [72].
Later studies used human induced pluripotent stem cells (hiPSCs) [73-75] as model systems, which may enable a personalized medical approach to treat CHD patients. With the power of CRISPR-Cas9, hiPSC-based models containing mutations corresponding to those identified in patients have begun to clarify the mechanisms underlying the pathogenesis of these diseases. In addition, mutations in cells derived from patients can be corrected using CRISPR-Cas9-mediated homology directed repair (HDR) [76]. The advantage of utilizing iPS cells edited by CRISPR-Cas9 is that they can be easily sequenced to confirm genomic editing and identify off-target effects.
1. Wolff-Parkinson-White syndrome
Wolff-Parkinson-White (WPW) patients suffer from cardiomyopathy due to ventricular pre-excitation, hypertrophy, ventricular tachyarrhythmia, heart failure, and sudden death. The onset of this syndrome can occur at any age, including in utero and early childhood [77,78]. Xie et al. [79] identified a novel mutation in the gene encoding Protein Kinase AMP-Activated Non-Catalytic Subunit Gamma 2 (PRKAG2) in familial WPW syndrome patients. Several mutations of this gene, which is responsible for AMP-activated protein kinase sensing of energy and nutrition, have shown to cause WPW syndrome. The authors identified a H530R missense mutation and confirmed this finding by establishing both transgenic and knock-in mice model that had abnormal cardiac electrical activity, ventricular hypertrophy, and excessive glycogen deposition. They subsequently applied an AAV9 mediated Cas9 and sgRNA delivery strategy to target the mutated PRKAG2 allele in their knock-in mice (+/H530R). A single intravenous injection of mixed AAV9 particles at postnatal day 4 successfully disrupted the mutant allele to restore left ventricular wall thickness and glycogen deposition. Consequently, they developed a potential CRISPR-Cas9 therapeutic strategy for WPW syndrome patients [79].
2. Duchenne muscular dystrophy
Duchenne muscular dystrophy (DMD) is an x-linked disease caused by mutations in the dystrophin gene, which is composed of 79 exons. This disease leads to progressive muscular weakness and severe cardiomyopathy [80,81]. Despite advancements in the care for DMD patients, there is no therapy that can reverse the course of this disease. The mdx mouse model of DMD (C57BL/10ScSn-Dmdmdx/J) contains a nonsense mutation in exon 23. Long and colleagues used a CRISPR-Cas9 strategy to correct this mutation [82]. They injected a Cas9 mRNA/sgRNA/ssODN (single-stranded oligodeoxynucleotide) template for HDR into mouse zygotes in the germ line to potentially correct this mutation in every cell. The authors examined 11 CRISPR-Cas9 corrected mice and 7 mice were confirmed to have a successful mutation correction via HDR. This strategy resulted in a mosaic correction ranging from 2% to 100%. The authors confirmed that the CRISPR-Cas9-mediated correction in mdx mice recovered from the DMD phenotype using histological analyses and serum creatine kinase measurements [82]. Because genome editing of a germ line condition is not currently feasible in humans, the authors presented an efficient delivery strategy for somatic cell correction of DMD mutations. Furthermore, El Refaey et al. [83] used another DMD mouse model (i.e., mdx/Utrp+/−) with AAV9-mediated systemic delivery of the Staphylococcus aureus Cas9/sgRNA for genome editing in the heart. In this study, approximately 40% of dystrophin expression was established in cardiomyocytes, which reduced cardiac fibrosis and improved contractility.
3. Holt-Oram syndrome
Holt-Oram syndrome is a rare congenital disease characterized by anterior pre-axial limb abnormalities and defective cardiac development, including septal defects [84-86]. The TBX5 gene has several mutational hotspots and is believed to be a causative gene in this syndrome [87,88]. At least 14 mutations (including null mutations and missense mutations) in human patients have been reported [84]. Using Zebrafish as a genetic model system, the importance of tbx5 during cardiogenesis has been shown in the heartstring knockout, which have a linear heart tube that fails to loop—mirroring developmental defects in human Holt-Oram syndrome [89]. Because of a gene duplication in this species, Zebrafish have 2 forms of tbx5, called a and b. Morpholino oligomers [90] were constructed to validate the observations from the heartstring and CRISPR/Cas9 knockout fish at the molecular and phenotypic levels. These experiments confirmed the vital importance of these genes in cardiogenesis. For the CRISPR/Cas9 knockout fish, the authors designed a sgRNA that targeted the start codon of tbx5b with Cas9 mRNAs, which was injected at the embryonic one-cell stage to create the F0 tbx5b knockout [91]. These fish showed alterations in somite size, vasculature cell differentiation, and subsequent trunk blood vessel patterning.
4. Heterotaxy syndrome
Heterotaxy syndrome is characterized by the left-right asymmetry defects that can affect the heart [92]. ZIC3, DNAH10, RNF15, and CFC1, have been reported as potentially causative genes. ZIC3 is a zinc-finger transcription factor belonging to the Gli superfamily that is located at the Xq26.2 chromosomal position [93]. Mutations in ZIC3 are associated with about 75% of x-linked, familial Heterotaxy syndrome patients with transposition of the great arteries and double-outlet right ventricles [11]. ZIC3 hypomorphic mice [94], Xenopus, and zebrafish models have been developed to validate the function of ZIC3 during early cardiogenesis [95,96]; however, there are currently no reports of CRISPR/Cas9 studies of this gene.
In addition to ZIC3, duplications of the DNAH10 gene located at 12q24.31 appear to be involved in Heterotaxy syndrome. DNAH10 is expressed in the cilia and the protein from this gene is a component of the inner dynein arm that connects to microtubules to enable flagellar motility in protozoans. The generation of CRISPR-Cas9-mediated dnah10 mutants in zebrafish (F0) showed cardiac defects reminiscent of Heterotaxy syndrome [97]. This study provided the first in vivo and functional evidence for dnah10 in left/right patterning. At the same time, the authors expanded the number of causative gene candidates for this syndrome by identifying RNF115 deletion mutations using CRISPR-Cas9.
5. CAKUTHED syndrome
A similar study was performed for the pre-B cell leukemia factor 1 (PBX1) gene mutation, which has been identified as a potentially causative gene in the Congenital Anomalies of Kidney and Urinary Tract Syndrome with or without Hearing Loss, Abnormal Ears, or Developmental Delay (CAKUTHED). Mutation of this transcription factor is associated with CAKUTHED combined with CHD. Alankarage et al. identified a novel missense mutation (R184P) in tetralogy of Fallot [98] patients [99]. To understand of this PBX1 mutation, the authors generated knockin animals using CRISPR-Cas9. These mice have some of the same developmental defects observed in CAKUTHED, such as a ventricular septal defect (VSD) [99].
6. Barth syndrome
Wang et al. [100] developed a hiPSC-based model of Barth syndrome. Mutations in the taffazzin (TAZ) gene can cause this syndrome, which is characterized by weakened cardiac muscle leading to dilated cardiomyopathy in male infants [100,101]. The authors discovered a mutation (T328C) in TAZ and established a cell model using 2 independent methods. In the first, the authors used hiPSC-derived cardiomyocytes (iPSC-CM) from a Barth syndrome patient. These cells were used to correct the T328C mutation using CRISPR-Cas9. In addition, the authors engineered the same mutation in a human iPSC line (PGP1-iPSC) by CRISPR-Cas9 targeting of exon 6 of TAZ. These independent cell models had a similar cellular phenotype to Barth syndrome patients, including abnormal cardiolipin biogenesis and mitochondrial function [100]. This research was followed up with a study using a mouse TAZ knockout mouse model and AAV9-mediated rescue to support the idea that TAZ is a critical target for the treatment of Barth syndrome [102].
7. Noonan syndrome
Mutations in the leucine zipper-like transcription regulator 1 (LZTR1) have been reported to cause the most common symptom of Noonan Syndrome—hypertrophic cardiomyopathy [103]. Noonan syndrome patients often suffer from pulmonary valvular stenosis, atrial and ventricular septal defects, in addition to hypertrophic cardiomyopathy and early disease onset [104]. Using hiPSCs to confirm phenotypic cardiomyopathy, an intronic mutation in LZTR1 was found to create a stop codon in the next exon. The CRISPR-Cas9 correction of this mutation restored expression of wild-type LZTR1 [103].
8. DiGeorge syndrome
DiGeorge syndrome is caused by a microdeletion of 22q11.2. This syndrome is characterized by developmental defects that include cardiovascular malformations [6-8]. Approximately 30 to 40 genes are deleted in DiGeorge syndrome. As a result, several causative genes have been studied. In particular, Tbx1 deletion induces haplo-insufficiency and cardiac defects similar to those observed in DiGeorge syndrome. CRISPR-Cas9 knockout of Tbx1 in murine stem cells (E14-Tg2a) enabled chromatin remodeling studies and transcriptome analyses to understand the pathogenesis of this syndrome [105]. Similarly, the putative adhesion receptor protein DGCR2, which has shown muscular defects as well as the risk for developing schizophrenia in DiGeorge syndrome, was deleted in mouse TT2 embryonic stem cells using CRISPR-Cas9 to validate and confirm the role of this protein in these processes [106-108].
9. Other CHD-related mutations
In nonsyndromic human CHD, several transcriptional factors are known to be required for proper cardiovascular development. Mutations in GATA4, TBX5, and MyH6/7 have repeatedly been reported as causative for CHD. For example, GATA4 mutations were identified in septal defects and conduction abnormalities [109,110]. Ang and colleagues developed a patient-specific, iPSC-based model from dermal fibroblasts to investigate one of these mutations. These authors also used CRISPR-Cas9 to correct the G296S mutation of GATA4 [109].
Gifford and colleagues performed exome sequencing of a 2-month-old infant with congestive heart failure with left ventricular noncompaction cardiomyopathy (LVNC) [111], which results from incomplete cardiomyocyte maturation and accounts for up to 10% of cardiomyopathies [112]. Three mutations were identified (i.e., A119S in NKX2-5, Q670H in MKL2, and L387F in MyH7 [111]. NKX2-5 governs cardiomyocyte development [113-115]. MLK2 is important in the epithelial-to-mesenchymal transition during embryonic development [116,117], and MyH7 encodes a crucial contractile protein. These mutations were confirmed in mice using CRISPR-Cas9 to create triple-heterozygous mice (Mkl2Q664H/+Myh7L387F/+Nkx2-5A118S/+), which recapitulated the LVNC phenotype. In addition, human fibroblast-derived hiPSCs harboring the same 3 mutations showed abnormal cardiomyocyte aggregation; thereby, supporting the claim that these 3 mutations are important in the pathogenesis of LVNC [111].
At the same time, MyH6 mutations can cause hypertrophic or dilated cardiomyopathy (HCM or DCM, respectively) and atrial septal defect (ASD) [118]. ASD type III (sinus venosus atrial septal defect) is also associated with MyHC6 mutations [119]. For instance, whole exome sequencing of 2 patients with CHD revealed a MyH6 mutation that caused truncation of α-myosin heavy chain protein that is implicated in ASD type III [120]. In addition, several other MyH6 mutations associated with CHD (mainly ASDs) have been confirmed using gain-of-function studies in the C2C12 cell line [121]. In the case of one MyH6 mutation (R443P) identified in a family with hypoplastic left heart syndrome [122], patient-derived hiPSCs were used in combination with CRISPR-Cas9 to demonstrate phenotypically-defective cardiomyocytes [123].
While all connective tissues can be affected Marfan syndrome, the most serious complication involves the heart. Mitral valve prolapse and aortic aneurysm are commonly associated with this syndrome, which results from mutations in the connective tissue scaffold protein, fibrillin-1 (FBN1) [124]. Correction of one of these mutations (T7498C) was tested using base editing 3 (BE3) [125]. Another example is LEOPARD (an acronym for Lentigines, Electrocardiographic defect, Ocular hypertelorism, Pulmonary stenosis, Abnormalities of the genitalia, Retarded growth and Deafness; and Noonan Syndrome with multiple lentigines) syndrome, which is a rare congenital disease with cardiac defects such as ventricular hypertrophy, cardiomyopathy, and abnormal electrical conduction [126-128]. As more mutations are identified in CHD patients [11], the possibility of utilizing CRISPR-Cas to correct these disease-causing mutations may soon be possible.
Conclusions
CRISPR-Cas9 is a versatile and robust tool for genome editing. The first clinical trial of this technology was launched in 2019 and is called the BRILLIANCE trial by Allergan (Dublin, Ireland) and Editas Medicine (Cambridge, MA, USA). This phase 1 and 2 trial uses CRISPR-Cas editing to restore vision in patients with Leber’s congenital amaurosis 10 (LCA10), which is caused by a point mutation in the CEP290 gene [129,130]. This trial relies in direct injection into the eye, near the photoreceptor cells, rather than systemic delivery or genome editing of cells removed from the body for subsequent replacement.
The application of CRISPR-Cas-mediated genome editing in humans requires very careful consideration, given that DSB is likely irreversible and certainly leads to genetic instability. The latter consequence has been shown in cancers and off-target DNA cleavage [60,131]. As a result, any genome editing would need to be both precise and efficient to ensure safety. In addition, patients would need to be closely monitored for immune reactions because of the bacterial origins of Cas enzymes are generally infectious in humans [34]. The relatively recent germline application of CRISPR-Cas9 editing in human pre-implantation embryos showed efficient genome editing with endogenous, germline-specific DNA repair to correct a paternal allele mutation in the MYBPC3, which causes cardiomyopathy [132]. Despite this success, many concerns remain; especially in regard to reproducibility for other heterozygous mutations and off-target DSBs.
In view of these concerns, somatic cell application of CRISPR-Cas is a safer choice, despite the challenge presented by inefficient Cas deliver to these cells. Modified AAVs are an attractive alternative; however, these viruses can be immunogenic and the most commonly used Cas9 protein from S. pyogenes, is above the packaging size limit for AAVs [133]. As a result, researchers continue to engineer and/or identify smaller versions of spCas9. While the promise of this technology cannot be disputed, Dr. Fyodor Urnov from the University of California, Berkeley, commented following the BRILLIANCE clinical trial that the "… technical challenges, and inherent safety concerns, are much greater." Therefore, further studies will be required prior to application of gene editing in patients with CHD.
Notes
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This work is supported by NRF-2020R1A2C1013377, NRF-2017R1D1A1B03030852 for H Seok and HL149401 from NIH to DZ Wang. We thank members of the Wang Laboratory for their careful reading and comments.