Modern diagnostic capabilities of neonatal screening for primary immunodeficiencies in newborns
Article information

Abstract
Population screening of newborns is an extremely important and informative diagnostic approach that allows early identification of babies who are predisposed to the development of a number of serious diseases. Some of these diseases are known and have effective treatment methods. Neonatal screening enables the early diagnosis and subsequent timely initiation of therapy. This helps to prevent serious complications and reduce the percentage of disability and deaths among newborns and young children. Primary immunodeficiency diseases and primary immunodeficiency syndrome (PIDS) are a heterogeneous group of diseases and conditions based on impaired immune system function associated with developmental defects and characterized by various combinations of recurrent infections, development of autoimmune and lymphoproliferative syndromes (genetic defects in apoptosis, gene mutation Fas receptor or ligand), granulomatous process, and malignant neoplasms. Most of these diseases manifest in infancy and lead to serious illness, disability, and high mortality rates. Until recently, it was impossible to identify children with PIDS before the onset of the first clinical symptoms, which are usually accompanied by complications in the form of severe coinfections of a viral-bacterial-fungal etiology. Modern advances in medical laboratory technology have allowed the identification of children with severe PIDS, manifested by T- and/or B-cell lymphopenia and other disorders of the immune system. This review discusses the main existing strategies and directions used in PIDS screening programs for newborns, including approaches to screening based on excision of T-cell receptors and kappa-recombination excision circles, as well as the potential role and place of next-generation sequencing technology to increase the diagnostic accuracy of these diseases.
Key message
· Neonatal screening for primary immunodeficiency diseases (PIDs) enables early diagnosis and subsequent initiation of therapy.
· Excision of T-cell receptors and kappa-recombination excision circles are cheaper alternative PID screening methods.
· Sanger DNA sequencing remains the reference method for detecting PID; however, next-generation sequencing technology is increasingly used to diagnose it.
· Here we developed a graphical algorithm for diagnosing primary immunodeficiency syndrome based on modern methods of screening for primary immunodeficiencies in newborns.
Introduction
In modern medicine, neonatal screening consists of a set of tests, examinations, or other easily used procedures designed to identify rare hereditary diseases. The testing process enables the identification and treatment of metabolic, genetic, endocrine, and hematologic disorders, many of which are potentially fatal conditions.
In different countries, neonatal screening is a priority public health program. Infants are screened for various nosological forms of the disease. In the Russian Federation, neonatal screening for 5 diseases is currently performed, including cystic fibrosis, phenylketonuria, galactosemia, adrenogenital syndrome, and congenital hypothyroidism. According to the order of the Moscow Department of Healthcare No. 935 on December 26, 2017, neonatal screening in metropolitan maternity hospitals has been expanded to 11 nosologies. It includes an additional 6 diseases from the group of organic acidurias, aminoacidopathies, and a group of fatty acid metabolism disorders (glutaric aciduria type 1, tyrosinemia type 1, leucinosis, methylmalonic/propionic aciduria, biotinidase deficiency, acyl-KoA-dehydrogenase deficiency of medium-chain fatty acids) [1].
In recent years, scientists and researchers from different countries have paid special attention to the problems of neonatal screening for primary immunodeficiency disease or primary immunodeficiency syndrome (PID/PIDS), that is associated with the steady increase in the incidence of this pathology and the need for its early diagnosis.
PIDS is a heterogeneous group of disorders of the immune system associated with defects in the immune system’s development and/or dysfunction. They are characterized by various combinations of recurrent severe coinfections, autoimmune reactions, the development of autoimmune and lymphoproliferative syndromes (genetic defects in apoptosis; mutation of the Fas receptor or ligand gene), granulomatous process, and malignant neoplasms. To date, 354 types of various disorders have been identified that are associated with 344 different gene defects. The clinical manifestations of PIDS are diverse, but many are associated with increased susceptibility to bacterial, viral, fungal, and mixed infections. This is based on defects in the functioning of various parts of the immune system.
Most often, severe combined immunodeficiency (SCID) cannot be diagnosed sooner than 6–8 months of life. Moreover, the delay in the diagnosis of other PIDS can be up to 5 or even more years from birth. This leads to the fact that more than 50% of children die before diagnosis and therapy initiation [2,3].
The average cost of an early bone marrow transplantation is 3 times lower ($120,000) than that of late transplantation ($360, 000) [4-6]. These data highlight the feasibility and importance of introducing neonatal screening for PID.
Flow cytometry, one of the most widely used methods for diagnosing PID, enables assessment of the immune system: specific cell populations and subpopulations, specific cell membrane, intracellular and intranuclear proteins, biological effects associated with specific immune defects, as well as certain functional immune characteristics Thus, it is a phenotypic analysis. However, despite the fact that flow cytometry is a sensitive and important tool for assessing immune system function and diagnosing PID, this method is targeted, which is why it helps to suspect PID only in the presence of certain symptoms, while these diseases cover several hundred various conditions affecting immune system development and/or functioning [7-9].
The clinical symptoms of PIDS are usually nonspecific and appear at different ages, which leads to significant diagnostic difficulties. The prerequisites for the diagnosis of PIDS are frequent recurrent infectious diseases of various types, more often with multiple localizations, moderate severity, and a severe course. In addition, such patients are characterized by a low clinical response to ongoing antibacterial and antimycotic therapy, the development of multiple autoimmune diseases or autoimmune syndrome, and the presence of a family predisposition to PIDS. Children with PIDS and general symptoms of gastrointestinal disorders are often identified at a gastroenterology appointment with complaints of malabsorption syndrome clinical manifestations, diarrhea, hepatomegaly/hepatosplenomegaly, and recurrent gastroenteritis [10]. In addition, children with PIDS often have infectious and inflammatory diseases of the osteoarticular system and anomalies or delays in bone and cartilage tissue formation (infectious arthritis, osteomyelitis, short stature, etc.) [11]. PIDS is characterized by the development of specific skin symptoms, including erythroderma, eczematous lesions, and subcutaneous abscesses [12]. Central nervous system dysfunction and the resulting neurological symptoms can range from mild cognitive disorders to serious disorders (ataxia, micro- or macrocephaly) [13]. In some cases, retinal damage may occur.
The effectiveness of treatment depends on the age at manifestation and on the timeliness of its appointment, which emphasizes the importance of early diagnosis and treatment to ensure better outcomes and cost effectiveness [14].
Conducting neonatal or selective screening makes it possible to detect these disorders with a high probability in the early stages before the onset of clinical symptoms.
T-cell receptor excision circles and kappa-recombination excision circles
T-cell receptor excision circles (TREC) and kappa-recombination excision circles (KREC) are PIDS screening methods that have been actively used in different countries in recent years. TREC is a marker that indicates changes in characteristics of the T-cell link of immunity. KREC indicates changes in the B-cell link of immunity TRECs are small round pieces of episomal DNA that are formed during the rearrangement of the T-cell receptor (TCR) in naive T-cells; thus, they are surrogate markers as they are found in the cells exported from the thymus but are absent in replicating peripheral blood cells [15,16]. The functional activity of the thymus decreases with age, which leads to a change in the amount of TREC in the peripheral blood cells [16]. However, the level of TREC in newborns is high, and as they grow older, the number of T-lymphocytes containing TREC decreases due to the expansion of peripheral T-cells [17].
Thus, the level of TREC in the peripheral blood of newborns reflects the activity of the thymus [18]. Quantitative studies have shown that approximately 70% of TCRD4 deletion rearrangements produce circular DNA. Therefore, the quantification of TREC has become a widespread, accurate, and noninvasive tool for detecting T-cell immune disorders [15].
TRECs were first visualized by electron microscopy as circular extrachromosomal DNA in mouse thymocytes back in 1982; later, it was demonstrated that they are a product of TCR rearrangement [17,19]. The TREC assay was developed by Douek et al. [18,20], who demonstrated that TRECs are specific for naive T-cells and described the age-related decline that occurs in healthy individuals.
In 2005, Chan et al. first described using the TREC test for large-scale screening of newborn infants for SCID and other forms of T-cell lymphopenia [21]. The TREC2 assay quantifies TRECs using real-time quantitative polymerase chain reaction (PCR) for DNA in a dry blood drop collected from newborns [22]. The maturation process of TCR3 consists of a random rearrangement of different DNA segments of variable TCR chains (α, β, γ, δ). Both coding and signaling compounds are created during rearrangements of the TCR-delta deletion from the TCR-alpha locus. During the analysis, TRECs are stable, not prone to degradation, and do not replicate with subsequent cell division, which makes them an ideal marker for naive T-cells [15]. Pilot studies showed that TREC had a 92.3% specificity and 100% sensitivity for diagnosing SCID; as of 2011, TREC testing was included as part of the newborn screening panel in several United States (US) states, including Wisconsin, Massachusetts, and California [23,24].
The first pilot study using the TREC method on PIDS was conducted in Wisconsin, USA, in 2008, led by Routes et al. [25]. Later that year, a child identified by newborn screening underwent a successful transplantation (Jeffrey Model Foundation, http://www.info4pi.org). Screening was subsequently introduced in Massachusetts, Louisiana, and New York in 2009, and California, Texas, and Pennsylvania in 2010. The number of cases diagnosed worldwide in 2018 was 94,024, 21.8% more than in 2013 [26]. More than 320 genes have been found to cause PIDS with a wide range of clinical phenotypes [26].
Wisconsin was the first US state to officially implement TREC for the detection of SCID in infants, and its screening program was the longest (44 months) recorded in the US [23]. A total of 71,000 children were examined, 8 of whom had T-cell lymphopenia of various origins. The Wisconsin program achieved a false-positive rate of only 0.03% in full-term infants and 0.14% in preterm infants [24]. In May 2011, 6 US states had already screened newborns for SCID using the TREC method. These programs identified 14 cases of classic SCID and 40 cases of T-cell lymphopenia that were not associated with SCID in a total of 961,925 infants. These data show that low or absent TREC in infants indicates any immune system disorder requiring follow-up tests [27].
The immune status is traditionally assessed by flow cytometry, a highly sensitive and important tool. Nevertheless, this method is quite expensive, is difficult to obtain, and requires appropriate training. TREC and KREC are cheaper alternatives for screening for and diagnosing PID. These methods can be used in small laboratories and rural areas where sophisticated and expensive tools are not available to conduct an initial PID assessment. The levels of TREC and KREC can also serve as indicators for flow cytometry analysis [8].
The TREC assessment method is currently included in neonatal screening in the US, Israel, New Zealand, Norway, Taiwan, some provinces of Canada, Switzerland, Germany, Iceland, Sweden, Italy (Tuscany), Spain (Catalonia), and some regions of Austria [28]. Criteria for newborn screening are based on World Health Organization recommendations. A retrospective analysis of 108 children in 2 hospitals in England showed a marked difference between treatment groups. In the group of 60 patients with an early start of treatment, there were 6 deaths (10%), while in the control group of 48 patients with a late start of treatment, there were 29 deaths (60%) [29]. Thus, the inclusion of TREC and KREC analyses in neonatal screening allowed a larger number of patients receive an early diagnosis and helped increase their survival rate and quality of life.
It is important to note that the study of the TREC and KREC methods was also performed in Russia [30]. In 2014–2016, the G.N. Speranskiy Children Hospital No. 9, Moscow, Russian Federation, together with the Dmitry Rogachev Federal Research and Clinical Center for Pediatric Hematology, Oncology and Immunology, conducted a pilot study to determine TREC and KREC amounts in patients from the Sverdlovsk region. Using the domestic multiplex test system, the contents of TREC and KREC were analyzed in samples of dry blood spots of 117 newborns, and PCR analysis showed that this system can reproducibly estimate the number of TREC molecules in model blood samples containing only 10% of the healthy children’s blood [31,32]. Thus, 10 μL of blood was sufficient for such an analysis. A similar experiment was performed for the KREC target. The system reliably identified the KREC target in 1% of normal blood, confirming the very good sensitivity of this assay: 5×103 TREC/KREC per mL and 100% specificity [31,33].
A retrospective study of TREC and KREC in blood samples of young infants who died of various causes suggested that some of the deaths were associated with the development of SCID and could have been avoided if this test had been introduced into neonatal screening programs [33-37].
Next-generation sequencing
Next-generation sequencing (NGS) is a DNA research technology for sequencing entire genomes or specific regions, including all 22,000 coding genes (whole exome) or a small number of individual genes.
In 2008, the first study was published on the genome sequence obtained using NGS. Until 2010, the genes responsible for PID development were identified only within the traditional Sanger sequencing method used alone or in conjunction with other genetic studies [38]. Sanger DNA sequencing remains the reference method for molecular diagnostics of PID; however, NGS is increasingly used to identify PID indication [39-42]. NGS techniques include whole-exome sequencing (WES), which can be used to detect mutations in genes encoding proteins and RNAs, and whole-genome sequencing for complex sequencing of the entire genome, including introns.
If it is necessary to study several candidate genes at once, NGS provides faster and cheaper diagnosis than Sanger sequencing, and NGS methods have already been used by several genetic laboratories for the diagnosis of various genetic disorders, including PID [42-45]. With the growing number of genes to be tested, especially for PID, Sanger sequencing becomes too expensive and ineffective [39], as it is targeted and requires an obvious candidate gene [46]. However, due to the lack of complete coverage of the exome, the error rate for WES is much higher than that for Sanger sequencing [47]. In addition, WES has a number of limitations, as only coding regions are amplified in this method.
Moreover, the overall sensitivity of this method for detecting complex structural changes (deletions, insertions, inversions, etc.) or repetitive sequences is low. Finally, if the gene of interest is in close proximity to pseudogenes with similar sequences (for example, the IKBKG and NCF1 genes), gene mis-mapping can occur, making it difficult to detect mutations [38].
There was a study conducted in Japan from November 2015 to April 2018 in which 2392 patients were genetically tested using NGS. 51 infants (2.1%, 51 of 2,392) were diagnosed with PIDS. Seven types of PIDS were found, and the most common (25 of 51, 49%) were combined immunodeficiencies with associated or syndromic symptoms. Thirty-five patients (68.6%) were cured or had improved outcomes after being diagnosed with PID.
According to the PID classification, 7 types of immunodeficiency diseases were identified in this study: 4 cases of immunodeficiencies affecting cellular and humoral immunity; 25 cases of combined immunodeficiencies with associated or syndromic features; 3 cases of predominantly antibody deficient; 6 cases associated with impaired immune regulation; 9 cases of birth defects related to the number or function of phagocytes; 3 cases of innate and innate immunity defects; and one case of an autoinflammatory disorder [48,49]. There were no cases of deficiencies in the complement system or phenocopies of innate immunity errors in the cohort. Forty-seven patients (47 of 1,680, 2.80%) with PID required intensive care, whereas only 4 (4 of 712, 0.56 %) did not.
TREC and KREC were performed on all 51 patients in this study. To confirm the results in positive patients, a subpopulation of lymphocytes was determined using flow cytometry. The results showed that 12 patients had decreased T-cell counts (<1,000 cells/µL) and 2 patients had decreased B-cell counts (<300 cells/µL). Among patients with reduced T-cell counts, 5 were diagnosed with DiGeorge syndrome, one with a CHD7 mutation, one with an ATM mutation, one with a CD40LG mutation, one with an IKBKG mutation, one with an IL2RG mutation, one with an LIG4 mutation, and one with a RAG1 mutation. Two patients with reduced B-cell counts harbored mutations. This result highlights that only 14 patients were identified with PID based on TREC and KREC levels detected in the pediatric group [49]. TREC/KREC neonatal screening is widely used for early PID diagnosis [50-54]. It plays a very important role in the identification of immunodeficiencies associated with T- and B-cell changes. However, this study showed that almost half of the patients had combined immunodeficiencies with associated or syndromic symptoms. Only one-quarter of the children had decreased T- or B-cells. Therefore, routine TREC and KREC screening may miss a PID diagnosis.
Thus, the data above demonstrate the clinical importance of genetic testing in children with early childhood infections. Given the rapid pace of improvement in NGS methods, it is possible that this technology will replace all currently known screening methods for PIDS in the future [38].
Based on modern methods of screening for primary immunodeficiencies in newborns, we developed a graphical algorithm for diagnosing this pathology. This algorithm will help neonatologists and pediatricians accurately and methodically diagnose primary immunodeficiencies in newborns and young children (Table 1).

Conclusion
There has been a steady increase in the number of newborns and young children suffering from various types of primary immunodeficiencies. Often, the late diagnosis of primary immunodeficiencies and untimely start of treatment lead to a serious disease course, significantly worsened prognosis, and increase mortality and disability rates in children. Therefore, scientists and researchers worldwide are now paying attention to neonatal screening for PID/PIDS.
PID and PIDS are a heterogeneous group of diseases and conditions of impaired immune system functions associated with developmental defects and characterized by various combinations of recurrent infections, the development of autoimmune and lymphoproliferative syndromes (genetic defect of apoptosis; mutation of the Fas receptor gene or ligand), granulomatous process, and malignancies. Modern advances in medical laboratory technologies already allow us to identify children with this pathology before the first clinical symptoms appear as well as children with severe forms of PIDS manifesting as T- and/or B-cell lymphopenia and other immune system disorders and begin therapy in the early stages of the disease, which significantly reduces mortality and disability and improves prognosis and quality of life. Our PID diagnostic algorithm facilitates and reduces the time to diagnosis (Fig. 1).
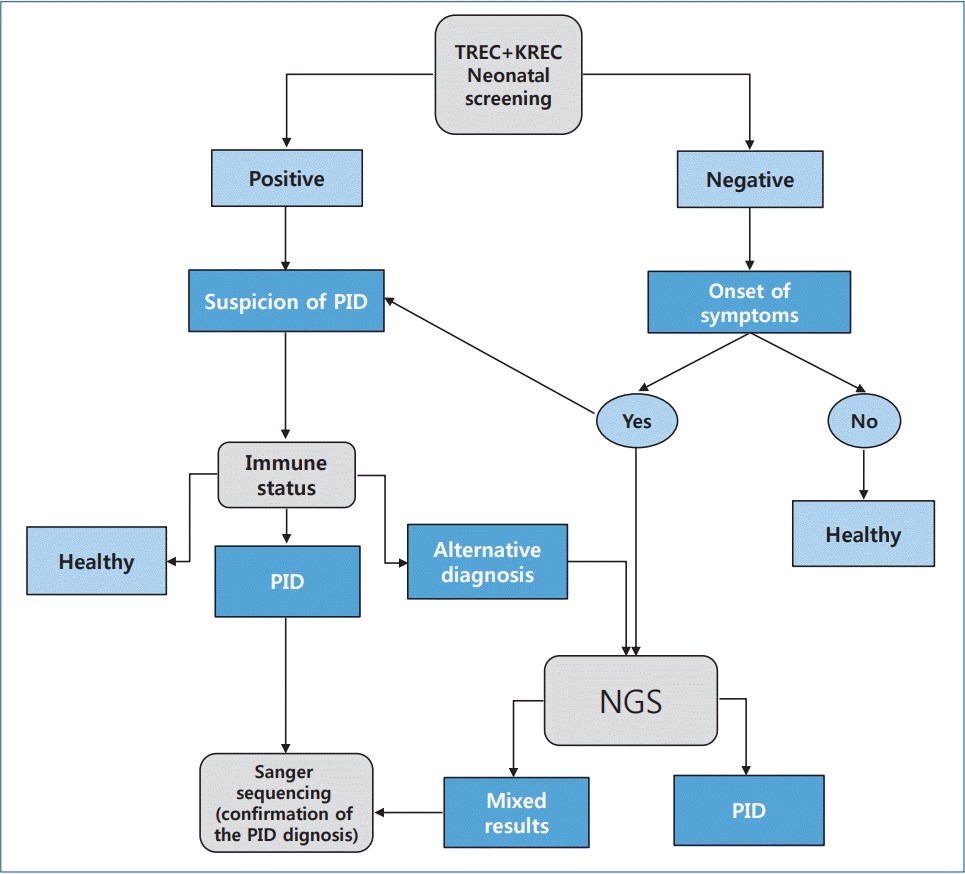
Algorithm of neonatal screening of primary immunodeficiencies in newborns. Concept of the best algorithm for neonatal screening for primary immunodeficiencies in newborns. TREC, T-cell receptors excision circles; KREC, kappa-recombination excision circles; PID, primary immunodeficiency diseases; NGS, next-generation sequencing.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.