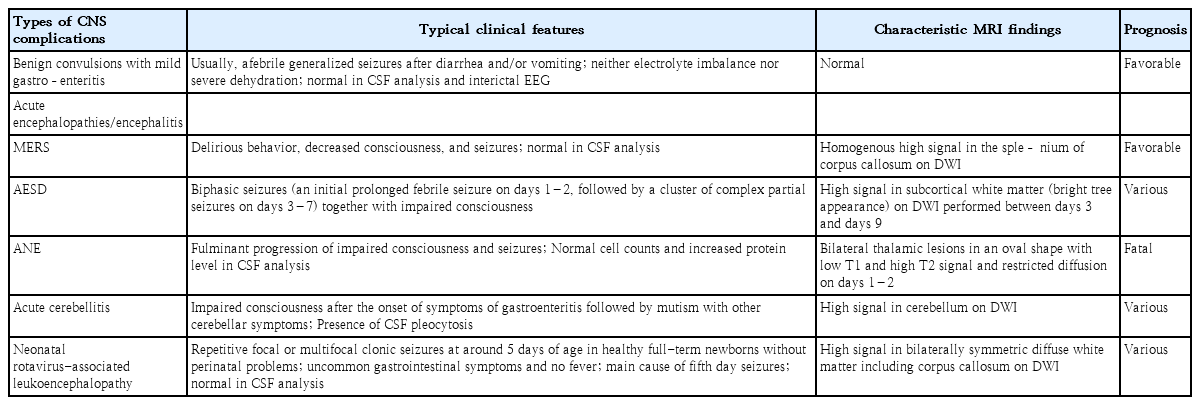
Rotavirus infection-associated central nervous system complications: clinicoradiological features and potential mechanisms
Article information

Abstract
Despite the introduction of vaccines in 2006, rotavirus remains one of the most common causes of pediatric gastroenteritis worldwide. While many studies have conclusively shown that rotavirus infection causes gastroenteritis and is associated with various extraintestinal manifestations including central nervous system (CNS) complications, extraintestinal manifestations due to rotavirus infection have been relatively overlooked. Rotavirus infection-associated CNS complications are common in children and present with diverse clinicoradiological features. Rotavirus infection-associated CNS complications can be classified based on clinical features and brain magnetic resonance imaging findings, particularly lesion location on diffusion-weighted imaging. Common clinicoradiological features of rotavirus infection-associated CNS complications include: (1) benign convulsions with mild gastroenteritis; (2) acute encephalopathies/encephalitis, such as mild encephalopathy with a reversible splenial lesion, acute encephalopathy with biphasic seizures and late reduced diffusion, and acute necrotizing encephalopathy; (3) acute cerebellitis; and (4) neonatal rotavirus-associated leukoencephalopathy. The precise mechanism underlying the development of these complications remains unknown despite a number of clinical and laboratory studies. Here we review the diverse clinicoradiological features of rotavirus infection-associated CNS complications and propose a hypothesis of their pathophysiology.
Key message
∙ Rotavirus infection-associated central nervous system (CNS) complications are fairly common in children.
∙ Common clinicoradiological features include benign convulsions with mild gastroenteritis, acute encephalopathies/encephalitis, cerebellitis, and neonatal rotavirus-associated leukoencephalopathy.
∙ Possible mechanisms for CNS complications include direct viral invasion into the brain via several potential routes such as the blood-brain barrier and vagus nerve, and entry of various brain-damaging mediators and activated immune cells into the brain.
Introduction
Rotavirus, one of the most common pathogens causing gastroenteritis among children under 5 years of age, is the leading cause of pediatric diarrhea-related deaths worldwide [1]. Before the introduction of vaccines, rotavirus gastroenteritis was responsible for the deaths of more than 500,000 children annually [2]. Since 2006, rotavirus vaccines have been introduced in more than 100 countries and significantly reduced the global burden of rotavirus disease [3]. During the first 10 years after vaccine introduction into the national immunization schedule for children, rotavirus disease–associated hospitalizations decreased by 67% (range, 18%–84%) and all-cause diarrhea-related deaths decreased by 42% (range, 3%–64%) [4,5]. In addition, rotavirus vaccination has reportedly decreased the hospitalizations for acute gastroenteritis-associated seizures in children <5 years old in several countries, including the US, Japan, the UK, Spain, and Korea [6-11]. Nevertheless, rotavirus gastroenteritis still causes more than 200,000 deaths annually [12]. Rotavirus was first reported in 1973 after the visualization of viral particles under an electron microscope in the duodenal mucosa of 9 children with acute nonbacterial gastroenteritis [13]. Rotavirus infection was initially considered confined to the gastrointestinal tract; however, many reports have shown that it may cause extraintestinal manifestations including various neurological disorders, hepatitis, cholestasis, pancreatitis, type 1 diabetes, respiratory illness, myocarditis, renal failure, and thrombocytopenia [14,15]. Rotavirus is a common cause of acute encephalopathy and encephalitis in children [16-19]. Since the first report of central nervous system (CNS) involvement with rotavirus in 1978 (one fatal Reye’s syndrome case, one encephalitis case) [20], a variety of rotavirus infection-associated CNS complications, such as convulsions with mild gastroenteritis [21-23], aseptic meningitis [24], mild encephalopathy with reversible splenial lesions [25-27], encephalitis [28,29], and cerebellitis [30], have been documented. Although rotavirus infection can involve CNS complications with various manifestations, such as impaired consciousness, seizures, and mutism, the underlying mechanisms by which rotavirus infection causes these complications are largely unknown.
This study aimed to examine the diverse clinicoradiological features and propose mechanisms of rotavirus infection-associated CNS complications based on previously published studies.
Clinicoradiological features of rotavirus infection-associated CNS complications
1. Benign convulsions with mild gastroenteritis
Benign convulsions with mild gastroenteritis are among the most common neurological manifestations related to rotavirus infection in children, occurring in approximately 1%–8% of patients with rotavirus gastroenteritis [14,31-34]. Although it has been documented worldwide, the number of reported cases is considerably higher in East Asian countries, particularly Japan, Korea, Taiwan, and China [21-23,35-37]/ This clinical condition is seen in previously healthy children aged 1 month to 6 years (peaking in 1–2-year-olds) and present with afebrile (or febrile) seizures that develop 1–6 days after gastroenteritis onset [34,35]. Some children experience seizures 12–24 hours before or even on the same day as the onset of diarrhea. Patients show neither electrolyte imbalance nor severe dehydration. Although seizures are usually the generalized tonic-clonic type, ictal electroencephalography has almost always demonstrated focal onset followed by secondary generalization [34,38]. Findings on interictal electroen cephalography, cerebrospinal fluid (CSF) analysis, and magnetic resonance imaging (MRI) are almost always normal (Table 1). Some patients show lesions in the splenium of the corpus callosum on diffusion-weighted imaging (DWI) [39]. Rotavirus and norovirus antigens are frequently identified in stool samples from these patients, and rotavirus ribonucleic acid (RNA) or antigens have also been detected in CSF samples in some cases [40-42]. Although patients commonly have multiple seizures, the outcome is favorable, with rare relapse, unimpeded development, and no epilepsy [34].
Diverse clinicoradiological spectrum of rotavirus infection-associated central nervous system (CNS) complications
2. Acute encephalopathies and encephalitis
As mentioned above, rotavirus commonly causes acute encephalopathy and encephalitis in children. In Japan, 2 national surveys were conducted of the epidemiology of pediatric acute encephalopathy during 2007–2010 and 2014–2017 [17,18]. Interestingly, rotavirus was ranked third among the identified pathogens of antecedent infection in acute encephalopathy in both surveys. Influenza virus and human herpes virus 6/7 placed first and second, respectively. In rotavirus-associated encephalopathy, the median age at onset was 2 years in both studies. A recent Swedish prospective cohort study also showed that rotavirus was the third most common pathogen implicated in childhood encephalitis [19]. In 61 of 89 cases (68.5%), the etiologies of encephalitis were established. The common viral etiologies included tick-borne encephalitis virus (n=13), enterovirus (n=10), and rotavirus (n=9).
In general, since viral infection-associated acute encephalopathy and viral encephalitis present with similar brain dysfunction such as acute onset altered consciousness and seizures, it is often difficult to distinguish them from each other; thus, the terms are sometimes used interchangeably in the clinical setting. Encephalitis typically shows inflammation of the brain parenchyma, while acute encephalopathy shows pathologically widespread noninflammatory brain edema that is either cytotoxic (cellular) or vasogenic (vascular) [43]. In principle, although the presence of CSF pleocytosis favors a diagnosis of encephalitis, an absence of CSF pleocytosis in patients with viral encephalitis is not uncommon [44]. On brain MRI, viral encephalitis tends to demonstrate asymmetrical focal or multifocal lesions as seen in herpes simplex virus encephalitis [45], while acute encephalopathy is disposed to show bilaterally symmetrical lesions [16]. Both conditions may reveal normal MRI findings.
Another nationwide survey of rotavirus-associated encephalitis/encephalopathy and sudden unexpected death conducted in Japan between 2009 and 2011 showed that, among a total of 58 cases of rotavirus-associated encephalitis/encephalopathy and 7 cases of sudden unexpected death, 36 patients (62.1%) with encephalitis/encephalopathy had no sequelae, while 15 (25.9%) had neurological sequelae and 7 (12.1%) had fatal outcomes [46]. Pleocytosis was observed in 9 of 40 patients (22.5%). Elevated lactate dehydrogenase levels (>500 IU/L) or acidemia (pH <7.15) were associated with poor prognosis.
Viral infection-associated acute encephalopathy is classified according to the pathogenic virus of antecedent infection, such as rotavirus-associated encephalopathy or influenza-associated encephalopathy. Alternatively, based on clinical manifestations and brain MRI findings, it can be classified as various syndromes, such as acute necrotizing encephalopathy (ANE), acute encephalopathy with biphasic seizures and late reduced diffusion (AESD), mild encephalopathy with a reversible splenial lesion (MERS), hemiconvulsion-hemiplegia-epilepsy syndrome, hemorrhagic shock and encephalopathy syndrome, Reye’s syndrome, and so forth, although unclassified acute encephalopathy is the most common [16]. According to the Japanese national surveys of 2007–2010 and 2014–2017, MERS was the most common syndromic diagnosis of rotavirus-associated encephalopathy (18 cases [45.0%]), followed by AESD (4 cases [10%]) and ANE (1 case [2.5%]) in the first study. The order was the same in the second survey as well: MERS (21 cases [46.7%]), AESD (5 cases [11%]), and ANE (2 cases [4.4%]) [17,18]. A full recovery was observed in 28 (70.0%) and 31 cases (68.9%), while neurologic sequelae were seen in 8 (20.0%) and 13 cases (28.9%) in the first and second survey, respectively. Three (7.5%) and 0 deaths (0%) were reported in the respective surveys. Here we describe the typical findings of MERS, AESD, and ANE.
1) Mild encephalopathy with a reversible splenial lesion
MERS typically presents with mild encephalopathy following prodromal symptoms such as fever, cough, vomiting, and diarrhea. Delirium (54%), decreased consciousness (35%), and seizures (33%) were the most common neurological symptoms [16,47,48]. On MRI, the splenium of the corpus callosum shows a homogenous high signal on DWI (Fig. 1A) [49]. T1 and T2 signal changes are mild without enhancement during the acute period. Reversible diffusion restriction in the splenium of the corpus callosum has been reported in various conditions, such as diverse infections, withdrawal of antiepileptic drugs, altitude sickness, Kawasaki disease, and hyponatremia [16]. A Japanese national survey showed that MERS was the second most common pediatric acute encephalopathy, accounting for 16% of cases [16]. Among MERS-related pathogens, rotavirus was ranked second (12%), following influenza virus (34%) [18]. The prognosis of MERS is generally favorable, with complete resolution regardless of treatment [17,47,48]. The high signal on DWI disappears within 2 months, leaving no atrophy [17].
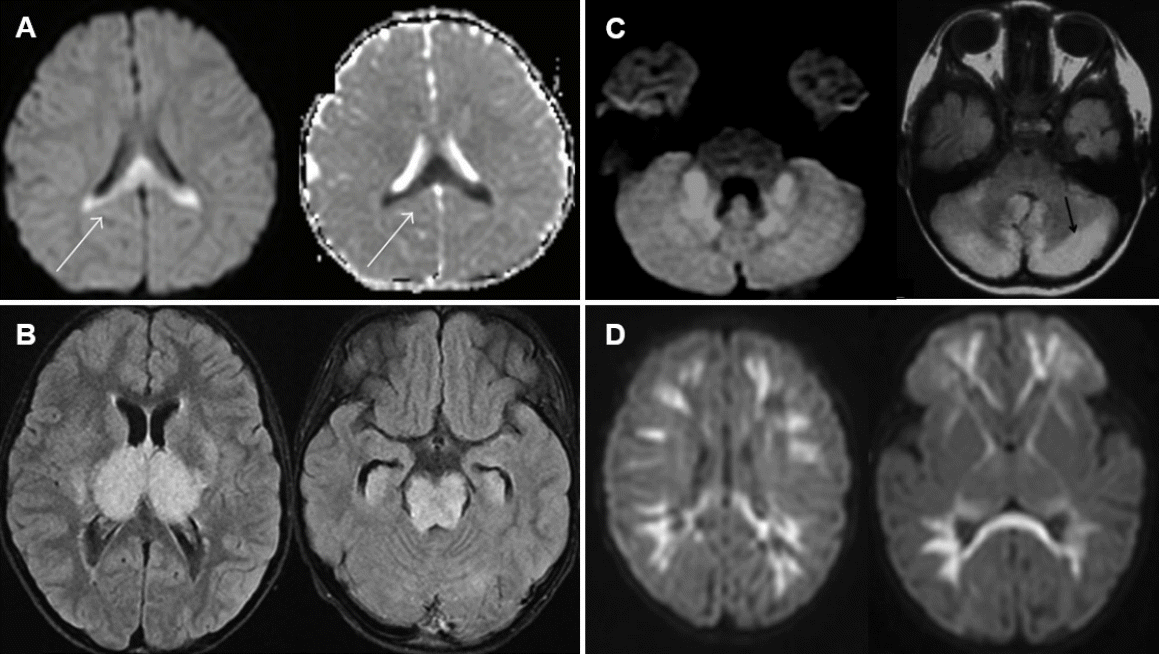
Characteristic magnetic resonance imaging findings of rotavirus infection-associated central nervous system complications. (A) Mild encephalopathy with a reversible splenial lesion. Note the diffusion restriction of the splenium in the corpus callosum (arrow). Reproduced from reference 49 (Copyright 2013, Hindawi Publishing Corp.). (B) Acute necrotizing encephalopathy. Note the hyperintensity of the thalamus in the oval shape, the basal ganglia, and the midbrain. Reproduced from reference 55 (Copyright 2020, Formosan Medical Association). (C) Acute cerebellitis. Note the diffusion restriction of the cerebellar peduncle and nuclei, and the high-signal intensity in the cerebellar cortex (arrow). Reproduced from reference 30 (with permission from American Society of Neuroradiology). (D) Neonatal rotavirus-associated leukoencephalopathy. Note the diffusion restriction of the bilateral diffuse cerebral white matter extending to the subcortical regions, corpus callosum, internal capsule, and posterior portion of the thalamus. Reproduced from reference 64 (with permission from Elsevier).
2) Acute encephalopathy with biphasic seizures and late reduced diffusion
AESD is clinically characterized by biphasic seizures, that is, an initially prolonged febrile seizure on days 1–2, followed by a cluster of complex partial seizures on days 3–7 in conjunction with impaired consciousness [16]. Between the early and late seizures, some patients (20%–30%) have normal consciousness with no neurological symptoms. MRI shows distinctive diffusion restriction in the subcortical white matter (bright tree appearance) on days 3–9 [48,50,51]. AESD has been predominantly reported in Japan, with an annual incidence of 100–200 cases [17,18]. The most common associated pathogens are human herpesvirus 6 (38%), followed by influenza virus (10%) and rotavirus (2%) [16]. DWI of MRI performed on days 1 and 2 usually shows no abnormalities, which may lead to an initial misdiagnosis of febrile status epilepticus. Clinical outcomes vary from normal to severe intellectual disability, paralysis, and epilepsy [52]. The mortality rate is relatively low (<5%). A recent study showed that a better prognosis was associated with a first seizure duration of less than 30 minutes and smaller MRI lesions (mainly in frontal areas) [52].
3) Acute necrotizing encephalopathy
ANE is characterized by the fulminant progression of impaired consciousness, seizures, bilateral thalamic lesions on MRI, and catastrophic outcomes [16]. ANE has been reported worldwide, but it appears to be more prevalent in East Asian countries such as Japan, Taiwan, and Korea [53-56]. Coma and seizures are very frequent, while vomiting and diarrhea are also common [16]. Shock may occur early after onset in severe cases. Bilateral symmetric thalamic lesions are a key feature of ANE. Thalamic lesions are oval-shaped (Fig. 1B) [16,55]. On days 1–2, the lesions showed low T1 and high T2 signal intensity accompanied by restricted diffusion. On day 3 and beyond, the thalamic lesions showed a concentric appearance due to T1 high-signal lesions in the central areas, indicating hemorrhagic changes. Apart from the thalamus, lesions are often observed in the periventricular white matter, internal capsule, putamen, upper brainstem tegmentum, and cerebellum. A CSF analysis shows normal cell counts and an increase in protein levels in two-thirds of patients [16]. Although an effective treatment for ANE is lacking, a retrospective study showed that steroid pulse therapy or intravenous dexamethasone within 24 hours after disease onset was associated with better prognosis in patients without brainstem lesions [57]. The reported mortality rate of ANE is approximately 30%, and most affected children die in the first week [58-61]. Survivors of ANE suffer from mild to moderate neurologic sequelae, and approximately 10% of patients recover completely [55,59].
3. Acute cerebellitis
Rotavirus cerebellitis is characterized by decreased consciousness 1–3 days after the onset of symptoms of gastroenteritis and subsequent mutism [30]. Consciousness clears between days 4 and 12, but mutism is observed after the recovery of consciousness. Mutism may last for up to 20 days. Other possible cerebellar symptoms include slow speech or dysarthria, hypotonia, ataxia, tremors, nystagmus, and dysmetria. MRI, particularly DWI, shows high-signal intensity in the bilateral dentate nuclei/cerebellar white matter in the acute and subacute phases, followed by increased signal intensity in the vermis and cerebellar cortex, and finally cerebellar atrophy 1–2 months after onset (Fig. 1C) [30,62]. Concomitant reversible splenial lesions are often observed during the acute phase [63]. CSF pleocytosis is frequently observed along with normal glucose and protein levels. Despite treatment with corticosteroids and intravenous immunoglobulins, many patients show mild to moderate cerebellar symptoms such as ataxia, dysarthria, tremors, and mental retardation [30].
4. Neonatal rotavirus-associated leukoencephalopathy
Neonatal rotavirus-associated leukoencephalopathy typically presents with repetitive or clustered focal or multifocal clonic seizures at around the 5th day after birth accompanied by bilaterally symmetric diffuse white matter and corpus callosum lesions on DWI [64]. We recently demonstrated that neonatal rotavirus-associated leukoencephalopathy was a main cause of fifth-day seizures, which prevailed in European countries and Australia between the 1970s and mid-1980s based on the similarity of clinical features and the annual incidence of an epidemic-like outbreak with abrupt disappearance [65]. Since the first report by our institute in 2014 [64], most cases of neonatal rotavirus-associated leukoencephalopathy have been documented in Korea, particularly in the confined southeast province of Korea, including Ulsan, Busan, and Jinju [66-68]. Between 2008 and 2016, in our institute, rotavirus-associated leukoencephalopathy accounted for 37.2% of neonatal seizures in full-term newborns, which peaked at 70.6% in 2012, gradually decreased, and finally disappeared in 2017. Seizures usually occur in healthy full-term newborns without a history of perinatal asphyxia. There is no known cause of neonatal seizures, such as hypoxic-ischemic encephalopathy or intracranial hemorrhage. Lesions on DWI are observed in symmetric diffuse patterns involving the cerebral white matter and corpus callosum. Lesions on DWI often extend to the internal capsule, external capsule, thalamus, basal ganglia, and brain stem following the white matter tracts (Fig. 1D) [64]. Only a small number of patients have gastrointestinal symptoms, such as diarrhea and vomiting, despite rotavirus infection. Almost all patients had no fever or rashes. Neither rotaviral RNA nor antigen was detected in CSF samples [66,68]. CSF pleocytosis was also not observed. Some patients showed a decrease in cerebral white matter volume and cerebromalacia in the white matter on follow-up MRI taken after 1–3 months [67]. Our previous study demonstrated that 18 of 32 patients (56%) with neonatal rotavirus-associated leukoencephalopathy had delayed performance on the mental and/or motor scales of the Korean Bayley Scales of Infant Development II assessment at a median age of 22 months [69]. The lesion extent on initial DWI was negatively correlated with the mental developmental scale.
Proposed potential mechanisms of rotavirus infection-associated CNS complications
The detection of rotavirus antigens and RNA in the CSF of patients with CNS complications supports the theory of direct viral invasion of the CNS [27,73-75]. However, CSF samples are not always positive for rotavirus antigen and/or RNA in cases of CNS complications [66,68]. Considering these observations, it is presumed that CNS complications may be caused by direct viral invasion of the CNS and/or circulating mediators with deleterious effects on the CNS. Here we review the structure of rotavirus, the histologic findings of rotavirus infection in enterocytes, and the role of rotavirus nonstructural protein 4 (NSP4), the most investigated protein in rotavirus infection at the molecular level. We subsequently propose the possible mechanisms of rotavirus infection-associated CNS complications as an overall entity rather than by each clinicoradiological type.
1. Rotavirus structure and classification
Rotavirus is a nonenveloped double-stranded RNA virus with a wheel-like icosahedral structure composed of 3 concentric capsids that surround a genome of 11 segments of RNA [1,76]. These RNA segments encode 6 structural viral proteins (VP1–4, VP6, and VP7) and 6 nonstructural proteins (NSP1–NSP6). Among the 6 structural proteins, surface glycoprotein VP7 and spike protein VP4 constitute the outermost layer and are responsible for eliciting an immune response. VP6 constitutes the middle capsid layer; based on the sequence and antigenic differences of VP6, rotavirus is classified into 10 different species (A–J) [77]. Species A rotavirus is the most common cause of infection in children [1]. Rotavirus is further classified into diverse genotypes based on sequence differences in RNA segments 7 and 4, which encode VP7 and VP4, respectively. VP7 and VP4 subtypes determine the G and P genotypes, respectively, in a dual classification system [76].
2. Histologic findings in rotavirus gastroenteritis
Rotavirus predominantly infects mature enterocytes at the top and middle of the intestinal villi, leading to the vacuolization of enterocytes [1]. Rotavirus infection and replication in the duodenal mucosa of infants cause shortening and atrophy of the villi, loss of microvilli, mononuclear cell infiltration, distended endoplasmic reticulum, and mitochondrial swelling in enterocytes [78,79]. The suggested mechanisms underlying these findings include virus-mediated apoptosis [80], NSP4-mediated mislocalization of the ZO-1 tight junction protein [81], binding to the basement membrane extracellular matrix proteins laminin subunit-β3 and fibronectin [82], and disruption of normal cellular homeostasis [83]. Previous in vitro studies demonstrated that rotavirus is able to infect neuronal cells, where NSP4 was detected [84,85]. Thus, rotavirus infection may cause a change in cellular structure and function, leading to cell death of neurons as well as enterocytes.
3. Rotavirus NSP4
Rotavirus NSP4 is a well-known viral enterotoxin that functions as a viroporin for the disruption of intracellular Ca2+ homeostasis through phospholipase C [1]. Viroporins are small virus-encoded ion channel proteins [86]. Rotavirus infection and NSP4 expression alone can increase cytoplasmic Ca2+ levels resulting from increased Ca2+ leakage from the endoplasmic reticulum via NSP4 viroporin [86,87]. The elevation of cytoplasmic Ca2+ levels is critical to the rotavirus replication process, including activation of autophagy, nucleation of rotavirus replication complexes called viroplasms, and assembly of the outer capsid protein VP7 [86,87]. NSP4 can also induce the production of nitric oxide and its metabolites. The overproduction of reactive nitrogen species is known to alter protein structure, thereby inhibiting their normal function, leading to DNA fragmentation and lipid oxidation [88]. One study showed that the treatment of intestinal cells with NSP4 increased inducible nitric oxide synthase mRNA expression [89]. Another study showed that the incubation of human intestinal epithelial cells with purified NSP4 caused NO2/NO3 accumulation in the incubation media [87]. In this study, 46 children with rotavirus infection showed higher urinary NO2/NO3 concentrations compared to their age-matched peers. Previous studies showed that inducible nitric oxide synthase activity depends on intracellular calcium levels and that NSP4 increases intracellular calcium levels. Thus, the authors interpreted that NSP4 increases NO production by elevating intracellular calcium levels. Rotavirus NSP4 can also elicit the production of proinflammatory cytokines, and a recent report demonstrated that NSP4 induces the secretion of proinflammatory cytokines such as interleukin (IL)-6 and tumor necrosis factor (TNF)-α together with nitrite from human and murine macrophage-like cell lines via Toll-like receptor 2 [90]. Another study showed that NSP4 induced the time-dependent generation of reactive oxygen species (ROS) in human intestinal epithelial cells [91]. Given that ROS can induce intestinal epithelial cell apoptosis in mice through Fas and Fas-L expression [92], NSP4 may induce cell apoptosis via ROS.
4. Possible routes of rotavirus invasion of the CNS
To enter the CNS compartment from the primary site of infection, neurotrophic viruses are known to follow 3 routes: (1) crossing the vascular endothelium; (2) accessing peripheral nerves; and (3) “Trojan horse” entry [93]. Rotavirus is also presumed to exploit the first 2 routes to reach the CNS (Table 2).

Proposed potential mechanisms of rotavirus infection-associated central nervous complications
1) Hematogenous spread via blood-brain and blood-CSF barriers and circumventricular organs
One of the most applauded mechanisms of viral invasion into the CNS is hematogenous spread across the vascular endothelium. Under normal conditions, the blood-brain and blood-CSF barriers are typically sufficient to protect the CNS from diverse insults including toxins, pathogens, and immune cells. Most blood vessels in the CNS are composed of nonfenestrated endothelial cells that have tight junctions that restrict the migration of cells and molecules through the interendothelial junction [94]. However, some neurotropic viruses use strategies that breach these barrier systems to enter the CNS. Several viruses, such as poliovirus [95], Epstein-Barr virus [96], and West Nile virus [97], directly infected human brain microvascular endothelial cell lines in vitro. Viral infection induces changes in endothelial cell properties, such as increased chemokine production, altered tight junction protein expression, and increased vascular cell adhesion molecule 1 expression [93]. These alterations potentially allow viruses to cross the vascular endothelium to reach the brain parenchyma. Under normal conditions, the choroid plexus epithelial cells act as a barrier between the systemic circulation and the CSF (blood-CSF barrier), with its predominantly tight junctions limiting paracellular diffusion, although the vascular endothelial cells at the choroid plexus are fenestrated and leaky [98-100]. Furthermore, hypoxia and inflammation disrupt barrier function [101]. A recent study showed that the Zika virus enters the CNS through the blood-CSF barrier instead of the blood-brain barrier in a mouse model [102]. The other potential doorway for hematogenous viral spread is the circumventricular organs in the brain, including the area postrema, vascular organ of the lamina terminalis, subfornical organ, pineal gland, and median eminence of the hypothalamus, which contain highly permeable capillaries unlike the rest of the brain [103].
Rotavirus antigenemia is common in children with rotavirus infection. In one study, antigens were detected in 22 of 33 serum samples (66.7%) from children with confirmed rotavirus infection versus none of the 35 samples from controls [104]. Surprisingly, another study showed that antigenemia was detected in 90% of children (51 of 57 cases) with rotavirus-positive stool and that infectious viruses were detected in 11 of 11 (100%) serum samples (viremia) from serum antigen-positive children [105]. Detection rates of rotavirus RNA in the blood range from 0% to 64% in rotavirus-infected children and are not always concordant with the presence of antigenemia [105].
Meanwhile, one study showed that rotavirus-specific proteins were identified in macrophages and B cells in gut-associated lymphoid tissue (Peyer’s patches and mesenteric lymph nodes) 2 days after the oral inoculation of murine rotavirus in mice [106]. Furthermore, these antigen-containing immune cells were identified in extraintestinal (inguinal) lymph nodes 7 days after inoculation. These findings suggest that the lymphatic system may act as a way for rotavirus to exit the intestine. Since the lymphatic system ultimately merges into the subclavian veins, rotavirus or its antigens may spread throughout the body through the systemic circulation.
Considering the high incidence of antigenemia and viremia in children with rotavirus infection, in conjunction with the isolation of viral antigens and RNA from the CSF in patients with CNS complications due to rotavirus infection, hematogenous spread across the blood-brain barrier, blood-CSF barriers, and capillaries of the circumventricular organs is a potential route of viral invasion.
2) Access via the peripheral nerves
Peripheral nerves are another potential pathway for viral entry into the CNS. Herpes virus type 1 is a representative virus that uses the strategy of peripheral sensory neuronal cell infection for CNS entry [93]. Once viruses enter the peripheral neuronal cells, they are moved between the cell body and the synaptic junction using 2 axonal transport systems: (1) the “anterograde transport system” exploits kinesin for movement from the cell body (soma) to the synapse, while (2) the “retrograde system” uses dynein for migration from the synapse back to the cell body [107]. After arriving in the CNS, viral dissemination within the CNS can occur through release into synaptic clefts (e.g., herpes simplex virus and rabies virus) or via microfusion with neighboring neurons (e.g., measles virus) [93]. Meanwhile, the gut has its own enteric nervous system that is uniquely organized with intrinsic microcircuits that facilitate the coordination of gastrointestinal function independent of the CNS [108]. The vagal nerve is known to play a role in communication between the gut and the brain. The vagal afferents, accounting for 80%–90% of all fibers, send the “up” signals from the gut to the brain, and the vagal efferents, accounting for 10%–20%, send “down” signals from the brain to the gut [109]. Additionally, one study showed widespread distribution of endoplasmic reticulum-associated rotavirus proteins, outer capsid protein VP7 and nonstructural protein NSP4 in cell bodies and dendrites of the dorsal root ganglion and hippocampal neuronal cells of Sprague-Dawley rats infected by plaque purified rhesus rotavirus [85]. Although there are no reports on rotavirus infection in the vagus nerve, given that rotavirus and its proteins have been identified within cell bodies, dendrites, and axons of the neuronal cells in vitro, it can be presumed that rotavirus in the intestine might initially infect the vagus nerve and then reach the CNS. Further investigations are required to identify whether rotavirus can infect the vagus nerve in vitro and in vivo.
5. CNS entry of circulating mediators produced by rotavirus infection
In addition to direct viral invasion into the CNS, the entry of various circulating mediators produced during rotavirus infection into the CNS can be considered a possible mechanism of CNS complications. Circulating mediators include proinflammatory cytokines, prostaglandins, ROS, nitric oxide metabolites, excitatory amino acids, and rotavirus antigens such as NSP4 [103]. Systemic inflammation caused by various stimulants, such as viruses, polyinosinic:polycytidylic acid (poly I:C, a mimic of viral double-stranded RNA), cytokines such as IL-1β, IL-6, and TNF-α, and bacteria and lipopolysaccharide, can damage blood-brain barrier integrity [110,111]. Thus, it is presumed that viremia or antigenemia, which often occurs in rotavirus infection as mentioned above, can cause systemic inflammation, thereby inducing a breach of the blood-brain barrier and blood-CSF barrier; eventually, the aforementioned deleterious mediators are able to enter the brain through the breach or the capillaries of the circumventricular organs.
Kawashima et al.112) showed that NO2 and NO3 levels in the serum and CSF of rotavirus gastroenteritis patients with convulsions were significantly higher than those in the control group. This result suggests the possibility of circulating NO metabolites entering the CNS. Host immune responses to rotavirus infection may also cause brain injury. Clinical studies have shown that rotavirus gastroenteritis is associated with increased levels of proinflammatory cytokines, such as IL-1β, IL-6, IL-8, and interferon gamma (IFN-γ) [14,113]. Newborns with rotavirus-associated leukoencephalopathy have significantly higher IL-6, IFN-γ, and monocyte chemoattractant protein-1 levels in the CSF than the control group [114,115]. Furthermore, CSF IL-6 levels correlate positively with white matter lesion extent. A previous report showed elevated PGE2 and PGF2 levels in the plasma and stool of rotavirus-infected children [116]. A clinical study showed that, in 19 children with CNS involvement associated with rotavirus infection, excitatory neurotransmitter levels (glutamate, glycine, and taurine) in the CSF were higher in patients with prolonged seizures accompanied by encephalopathy than in patients with a brief cluster of seizures without encephalopathy [117].
As mentioned earlier, viral encephalitis tends to demonstrate asymmetrical focal lesions on MRI as in herpes simplex virus encephalitis, while rotavirus-associated encephalopathy is generally characterized by bilateral symmetry, the predilection of cerebral white matter including the corpus callosum, and the lesions are well-demarcated, particularly on DWI. Considering these distinct features, it is likely that rotavirus-associated CNS complications may be attributed to the combined effects of CNS entry of various mediators and immune cells as well as virus and viral antigens across blood vessels in the brain rather than virus-induced cytopathic effects that developed after the direct viral infection of neurons and glial cells.
6. CNS entry of activated circulating leukocytes
Systemic inflammation caused by a viral infection can induce leukocytes in the blood vessels to transmigrate across the blood-brain barrier, and leukocytes to reach the brain can injure neuronal cells. The presence of CSF pleocytosis in patients with acute rotavirus-associated encephalopathies/encephalitis and cerebellitis indicates the recruitment of leukocytes from circulation into the CNS [30,46]. The migration of leukocytes from the vascular lumen to the infected sites engages sequential adhesive molecular interactions between leukocytes and endothelial cells [118-121]. When viruses infect cells, various chemokines are produced and bind to their cognate chemokine receptors on circulating leukocytes, thereby activating leukocyte integrins, such as very late antigen 4 and lymphocyte function-associated antigen 1, to cluster outside the cell membrane [118]. Besides, vascular endothelial cells enhance the expression of cell adhesion molecules such as vascular cell adhesion molecule 1 and intercellular adhesion molecule 1, which are clustered in the luminal side of vessels after a viral infection [119]. Binding between integrins of circulating leukocytes and cell adhesion molecules on vascular endothelial cells leads to activation of the endothelial cell signaling pathway, which elicits opening of the tight and adherence junctions, and stress fiber formation (endothelial contraction) via actin remodeling [120,121]. These microstructural alterations facilitate passage of the leukocytes through the vascular endothelial cells (diapedesis) and eventual arrival at the inflamed site. Leukocytes that reach the brain can injure the neurons and glial cells.
7. Considerations for future studies of the pathophysiology of rotavirus-associated CNS complications
In future research on the pathophysiology of rotavirus-associated CNS complications, the following should be considered. First, if rotavirus itself, its proteins and RNA, harmful mediators, and immune cells could induce brain lesions, what determines the specific lesion location? Second, what causes bilateral symmetry in brain lesions in almost all rotavirus infection-associated CNS complications? Third, why is cerebral white matter predominantly injured in most rotavirus infection-associated CNS complications instead of cerebral gray matter, as seen in MERS, AESD, and neonatal rotavirus-associated leukoencephalopathy? Finally, what other possible mechanisms are at play in different types of rotavirus infection-associated CNS complications?
Conclusion
Although rotavirus is a well-known most common cause of pediatric gastroenteritis worldwide, rotavirus infection-associated CNS complications have been relatively overlooked. However, rotavirus infection-associated CNS complications are fairly common in children and their clinicoradiological features are diverse. These have been classified mainly on the basis of the clinical features and characteristics of brain MRI, particularly lesion location on DWI. Common clinicoradiological features of rotavirus infection-associated CNS complications include benign convulsions with mild gastroenteritis and acute encephalopathies/encephalitis such as MERS, AESD, ANE, acute cerebellitis, and neonatal rotavirus-associated leukoencephalopathy. Despite a variety of clinical and laboratory studies, the precise mechanisms underlying these complications remain unknown. The direct viral invasion into the brain via several potential pathways, such as the blood-brain and blood-CSF barriers and vagus nerve, and the entry of activated immune cells and various brain-damaging mediators into the brain through the breach of blood vessels in the brain, are potential mechanisms for rotavirus infection-associated CNS complications. Further investigations are needed to elucidate the pathophysiology of each type of rotavirus infection-associated CNS complications. Moreover, proper treatment strategies should be identified according to the pathophysiology.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.