Development of orphan drugs for rare diseases
Article information

Abstract
Most rare diseases (orphan diseases) still lack approved treatment options despite major advances in research providing the necessary tools to understand their molecular basis and legislation providing regulatory and economic incentives to expedite the development of specific therapies. Addressing this translational gap is a multifaceted challenge, a key aspect of which is the selection of an optimal therapeutic modality to translate advances in rare disease knowledge to potential medicines known as orphan drugs. There are several strategies for developing orphan drugs for rare genetic disorders, including protein replacement therapies, small-molecule therapies (e.g., substrate reduction, chemical chaperone, cofactor, expression modification, and read-through therapies), monoclonal antibodies, antisense oligonucleotides, small interfering RNA or exon skipping therapies, gene replacement and direct genome-editing therapies, mRNA therapy, cell therapy, and drug repurposing. Each strategy has its own strengths and limitations in orphan drug development. Furthermore, numerous hurdles are present in clinical trials of rare genetic diseases because of difficulty with patient recruitment, unknown molecular physiology, the natural history of the disease, ethical concerns regarding pediatric patients, and regulatory challenges. To address these barriers, the rare genetic diseases community, including academic institutions, industry, patient advocacy groups, foundations, payers, and government regulatory and research organizations, must become engaged in discussions about these issues.
Key message
· Orphan disease is a rare disease, primarily affecting newborn and children. Vast majority of orphan diseases has genetic background.
· Orphan disease is individually rare. But as a whole, it is not rare, becoming a great socioeconomic burden.
· The diagnosis of rare genetic disease has been problematic, but recent progress of genome analysis technologies makes it faster and more precise.
· There are many unmet needs as to the curative treatment. However, the number of treatable rare diseases is growingly increasing owing to the development of biotechnology.
· Most orphan drugs are extremely expensive because of numer ous hurdles during the process of drug development as well as small number of patients.
Introduction
Orphan diseases are rare conditions affecting only a small proportion of the population. They are called “orphan” diseases because they are often neglected by pharmaceutical companies and researchers, as they do not generate enough revenue to justify the cost of developing treatment strategies. Over 7,000 orphan diseases affect an estimated 350 million people worldwide. Approximately 6,000–8,000 rare diseases have been identified to date, 80% of which are caused by mutations in numerous genes. Thus, a significant proportion of orphan diseases are genetic disorders. However, it is important to note that all the genetic causes of orphan diseases are not yet completely understood. Some genetic mutations may be rare and have yet to be identified, and some genetic causes of orphan diseases may be multifactorial and involve a combination of genetic and environmental factors.
Orphan drugs are medications or therapies that are used to treat rare diseases, also known as orphan diseases. The term “orphan drug” is used because these medications often do not have a significant market or patient population, which makes their development less profitable for pharmaceutical companies. Orphan drugs are typically designated by regulatory agencies such as the U.S. Food and Drug Administration (FDA), European Medicines Agency (EMA), and Korean Ministry of Food and Drug Safety (MFDS) as treatment approaches for diseases that affect less than 200,000 people in the United States, less than five in 10,000 people in the European Union (EU), or less than 20,000 people in South Korea [1]. The designation of orphan drug provides incentives such as tax credits, grants, and fee waivers for pharmaceutical companies to develop treatment strategies for rare diseases. The exact number may vary depending on the source. However, more than 600 orphan drugs for over 800 orphan diseases have been approved by the FDA (Fig. 1), and over 150 orphan drugs have approved by the EMA for treating rare diseases. The market for orphan drugs for rare genetic disorders is substantial and is expected to continually grow. According to a market research report, the global orphan drug market was valued at about $200 billion in 2020 and is projected to reach about $400 billion by 2030, growing at a compound annual growth rate of approximately 12% from 2020 to 2030 [1-4].
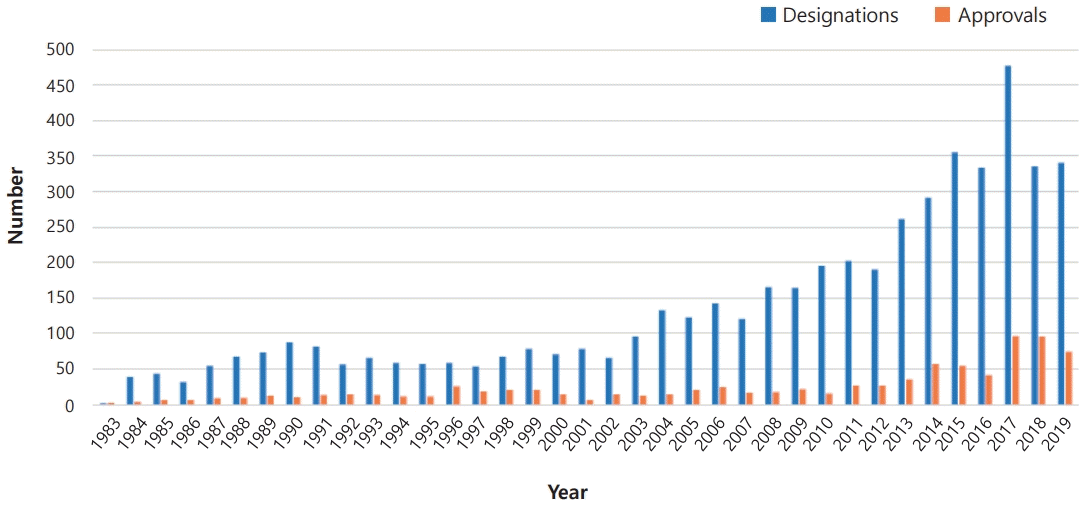
U.S. Food and Drug Administration orphan drug designation and total approvals by year, 1983–2019. The blue and red bars represent the number of approvals and designations, respectively.
Socioeconomic burden of rare genetic disorders
1. Economic cost
According to data from the IQVIA Institute for Human Data Science, global expenditure on orphan drugs exceeded $200 billion in 2020, and it is projected to continue to increase in the coming years. This is partly because of the high cost of individual orphan drugs, which can sometimes reach hundreds of thousands of dollars per patient per year [5].
2. Hospital and healthcare system burdens
Hospitals and healthcare systems face considerable burdens with regards to the treatment and management of patients with rare diseases. High costs strain hospital budgets and resources, and patients with rare diseases often require complex and extensive critical care, which can be difficult for hospitals to provide owing to relatively small number of patients and limited specialized expertise. Rare and ultrarare diseases can be challenging to diagnose because of their low incidence and complex symptoms, placing considerable burden on hospital resources. Owing to the small patient population and little financial incentives, the availability of specialized care may be limited. Patients with rare disorders often require care from multiple healthcare providers and specialists such as neonatal intensivists, placing substantial burdens on the hospitals coordinating and managing their care. Rare genetic diseases are becoming increasingly recognized, and their burden on the healthcare system is evident from their high mortality rate, disability, years of life lost, rate of admission or readmission to diverse hospital settings (general pediatric floor, neonatal intensive care unit, pediatric intensive care unit), rate of admission to long-term care and comfort care settings, and cost of illness [1,6,7].
3. Unmet needs
Diagnosing rare genetic disorders remains challenging for patients, doctors, and healthcare systems owing to insufficient characterization of the natural history of several rare diseases. Thus, majority of patients and their parents undergo diagnostic odyssey, which can be a long and exasperating journey for patients and their families seeking an accurate diagnosis. Typically, it can take up to 6 years from symptom onset to a correct diagnosis, during which time patients undergo an average of 14 diagnostic procedures and receive 4.5 diagnoses [8]. Approximately 4,400 genes are currently known to cause 6,300 rare genetic phenotypes (Online Mendelian Inheritance in Man), and recent data suggest that over 9,000 single-gene phenotypes might ultimately be discovered and molecularly defined. Nextgeneration sequencing, including whole-exome and whole-genome sequencing, is an established diagnostic tool used to reveal the genetic background of rare genetic disorders. However, its diagnostic yield is currently less than 50%. Recent advances have made these technologies both affordable and indispensable [9].
An extremely low percentage of rare genetic diseases is currently curable with orphan drugs. Although a few orphan drugs have been developed to treat rare genetic diseases, many remain incurable. According to some estimates, FDA-approved treatments are available for less than 5% of rare genetic disorders, although this number may vary depending on the specific populations and data sources. Additionally, an increasing number of drugs are currently being developed for rare genetic disorders, but hardly a small fraction of the total number of rare genetic disorders has been targeted. Developing orphan drugs can improve a pharmaceutical company’s reputation by demonstrating its willingness to invest in treating rare and often neglected diseases. Several pharmaceutical companies consider the development of orphan drugs a way to diversify their product portfolios and ensure long-term sustainability [10-12]. The development of drugs for rare genetic disorders is a complex and challenging process, and not all companies can do so successfully. Moreover, the prices of orphan drugs are relatively high compared to those of other drugs [13].
Orphan drug development strategies (Table 1) [14-16]

Strategy and examples of the diseases targeted for orphan drug development
1. Protein replacement therapy
Enzyme replacement therapy (ERT) is used to treat lysosomal storage diseases (LSD) caused by lysosomal hydrolase deficiency. ERT involves the introduction of a functional enzyme into the body to compensate for deficient or missing enzymes. ERT was first approved in the 1990s for Gaucher disease, and the first ERT drug replaced placentaderived alglucerase, with imiglycerase in Chinese hamster ovary cells expressing recombinant human-β-glucocerebrosidase. Subsequently, ERT drugs have been developed and approved for more than 10 LSD, including Fabry, Pompe, mucopolysaccharidosis types I,II,IVA, VI, VII, and cholesteryl ester storage diseases (Table 2) [17,18]. In addition, LSD and ERT have recently been used in hypophosphatasia with recombinant human bone-targeted tissue-non-specific alkaline phosphatase [19]. Pegvaliase is an FDA-approved ERT used in adults with phenylketonuria (PKU) and uncontrolled blood phenylalanine concentrations [20]. Coagulation factor replacement therapy is a treatment for hemophilia A and B. Recombinant factors VIII and IX are replaced to compensate for the deficiencies. Novel therapies for hemophilia are currently available, which can be administered via subcutaneous injection, to prolong the drug’s half-life or bypass the inhibitors. Another example is von Willebrand disease, which is caused by deficiency or dysfunction of the von Willebrand factor, a protein involved in blood clotting [21].

Current status of approved enzyme replacement therapies for rare diseases
2. Small-molecule therapy (Fig. 2)
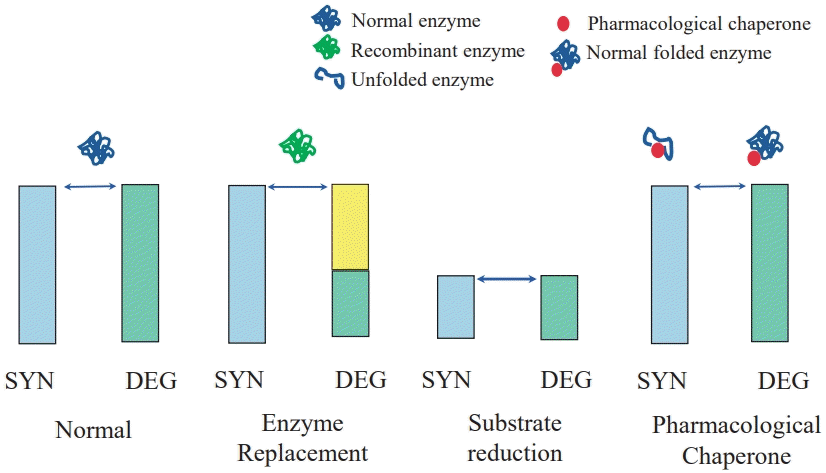
Molecular mechanism of small-molecule therapy in lysosomal storage diseases. SYN; synthesis; DEG, degradation.
Small-molecule therapies have several advantages and disadvantages. Small molecules are typically smaller than biologics and can be easily synthesized in a laboratory. This makes small-molecule therapy less expensive and easier to scale up for commercial use. Small molecules can easily cross cell membranes (such as the blood–brain barrier) and target intracellular proteins and enzymes, making them useful for treating diseases caused by mutations in intracellular signaling pathways. Small molecules can also be administered orally, which is convenient for patients and improves adherence. However, small molecules are often not as specific in their targeting as biologics; moreover, they may have off-target reactions that can cause side effects. Small molecules are metabolized or excreted quickly, limiting their effectiveness and requiring frequent dosing [16].
1) Substrate reduction therapy
Substrate reduction therapy (SRT) is a treatment for certain rare genetic disorders that involve the accumulation of toxic substances in the body. These disorders are caused by deficiencies in enzymes that break down the toxic substances, leading to their accumulation and disease symptoms. SRT aims to reduce the levels of toxic substances in the body and restore metabolic homeostasis by limiting the amount of substrate synthesized (and eventually subjected to catabolism) to a level that can be effectively cleared by the impaired enzymes. Therefore, it alleviates the disease symptoms by inhibiting the synthesis of the toxic substances rather than introducing a functional form of the missing enzyme as in ERT. A well-known example is nitisinone, a hydroxyphenylpyruvate dioxygenase inhibitor used as an adjunct to dietary restrictions in treating hereditary tyrosinemia type I. Another example is Gaucher disease, which is treated by the oral administration of the SRT drug eliglustat, a specific inhibitor of glucosylceramide synthetase approved by the FDA in 2014. Additionally, Niemann-Pick disease type C (NPC), a rare autosomal recessive lipid storage disorder characterized by impaired intracellular lipid trafficking and progressive neurological symptoms leading to premature death, is also an example of the disease treated using SRT. NPC results in a wide range of symptoms, including intellectual disability, ataxia, and hepato- and splenomegaly. Miglustat, a small iminosugar molecule that reversibly inhibits glycosphingolipid synthesis, is the only disease-specific drug approved for treating progressive neurological manifestations of NPC in adults and children. A few central nervous system–accessible inhibitors (such as lucerastat and ibiglustat) recently demonstrated promising preclinical and pharmacokinetic results for Fabry and type 3 Gaucher diseases [17,22].
2) Chemical chaperone therapy
Chemical chaperone therapy treats certain rare genetic diseases caused by the misfolding or misprocessing of proteins, which leads to their accumulation and occurrence of disease symptoms. Chemical chaperone therapy corrects the protein misfolding or misprocessing, thereby improving protein function and alleviating disease symptoms. This is achieved by using small molecules called chemical chaperones that stabilize the misfolded or misprocessed proteins and facilitate their proper folding, processing, and function. An example of a disorder treated with chemical chaperone therapy is Fabry disease, an X-linked lysosomal multisystem storage disorder caused by an alpha-galactosidase A (GLA) gene mutation that results in reduced activity or deficiency of alpha-galactosidase A. This leads to the storage of intracellular globotriaosylceramide in vital organs, including the kidneys, heart, and nervous system. Intravenous ERT has been the established treatment for 20 years.
Oral chaperone therapy was recently introduced as a therapeutic alternative for patients with amenable mutations. Migalastat was approved by the FDA in 2018 [23]. Another sample disease is cystic fibrosis (CF), a genetic disorder caused by mutations in the CFTR gene that result in an improperly folded and processed protein, leading to its accumulation and disease symptoms. Chemical chaperone therapy for CF of a specific genotype involves the use of small molecules that stabilize the CFTR protein,facilitate its proper folding and processing, improve its function, and alleviate the symptoms associated with the disease. Pharmacological chaperones (such as lumacaftor/ivacaftor) that bind directly to F508del-CFTR and correct its mislocalization are promising therapeutics for CF approved by the FDA in 2018 [24]. However, to date, it has only slightly improved lung function. Patients with chronic neuronopathic Gaucher disease may benefit from combination of ERT and chaperone therapy with ambroxol [25]. Chemical chaperone therapy for Pompe disease uses small molecules that stabilize the alpha-glucosidase protein, facilitate its proper folding and processing, improve its function, and alleviate the symptoms associated with the disease. Numerous studies are currently ongoing on target diseases that utilize pharmacological chaperone therapy [16,26].
3) Cofactor therapy
PKU is a rare genetic disorder caused by mutations in the PAH gene, which encodes the enzyme phenylalanine hydroxylase. Sapropterin dihydrochloride is a small molecule that can increase the activity of the enzyme, thereby reducing the levels of phenylalanine in the body and preventing the neurological complications of PKU [27]. Cofactor therapy is effective for many inborn errors of metabolism, such as biotin therapy in holocarboxylase synthetase deficiency, biotinidase deficiency, vitamin B12 in methylmalonic acidemia, and vitamin B6 in homocystinuria [28].
4) Expression modification therapy
Gene expression modification using small-molecule therapy is used to treat certain rare genetic disorders that involve the regulation of gene expression. This approach uses small molecules to modify the expression of specific genes, leading to changes in cellular function and the alleviation of disease symptoms. One example of a rare genetic disorder that can be treated with gene expression modification using small-molecule therapy is sickle cell anemia, which is caused by a mutation in the HBB gene, which encodes the β-globin subunit of hemoglobin. Hydroxyurea, approved by the FDA for treating sickle cell anemia, is a small molecule that increases the production of fetal hemoglobin and reduces the sickling of red blood cells.Its major mechanism of action involves the inhibition of ribonucleotide reductase (RR), the enzyme involved in transforming ribonucleosides into deoxyribonucleosides for DNA synthesis. Hydroxyurea is a potent RR inhibitor that reduces intracellular deoxynucleotide triphosphate pools and acts as an S-phase-specific agent that inhibits DNA synthesis [29].
5) Read-through therapy
Read-through therapy is a therapeutic approach for treating rare genetic disorders caused by premature stop codons, also known as nonsense mutations. These mutations are caused by a single change in the DNA sequence that prematurely arrests protein translation, resulting in a truncated, nonfunctional protein. The read-through therapy aims to overcome the premature stop codon by using small molecules that bind to the ribosome and allow it to “read-through” the premature stop codon, resulting in the production of a full-length functional protein. This approach can potentially restore the function of affected protein and treat the underlying genetic disorders. Ataluren, a drug under development, is used to treat Duchenne muscular dystrophy (DMD), which is caused by a specific type of nonsense mutation. However,this approach has not yet been approved [30].
3. Antibody therapy
Biological therapeutic strategies have advantages and disadvantages. Biologics such as monoclonal antibodies feature specific targeting and can bind to specific molecules or receptors on cells, making them useful in treating diseases caused by mutations in cell surface signaling pathways. Biologics have longer half-lives and may require less frequent dosing. Biologics can also target intracellular proteins and enzymes; however, they are typically larger molecules and are more difficult and expensive to produce. It is generally difficult for biologics to penetrate the blood–brain barrier; therefore, disorders involving the central nervous system are not treatable, and biologics are typically administered parenterally, such as intravenously or subcutaneously, which can be inconvenient for patients. Biologics are more likely to induce an immune response, which can limit their effectiveness over time [16,31].
Monoclonal antibodies are laboratory-made proteins that mimic the immune system’s ability to fight off harmful pathogens. These proteins can be used to target specific molecules, such as proteins or receptors, which are involved in disease occurrence. Monoclonal antibody therapy can effectively treat rare genetic disorders by blocking the function of disease-causing proteins or stimulating the immune system to attack the diseased cells. Burosumab is a monoclonal antibody that targets and inhibits fibroblast growth factor 23 (FGF23) activity.Its effectiveness has been proven in X-linked hypophosphatemia, in which FGFR23 is highly elevated, leading to renal phosphate wasting [32]. Eculizumab, a monoclonal antibody against paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome, was approved by the FDA in 2007, and its estimated annual cost is $500,000–$700,000 [33]. Familial hypercholesterolemia is a rare genetic disorder caused by mutations in the LDLR, APOB, or PCSK9 genes, all of which are involved in cholesterol metabolism. A rare cause of familial hypercholesterolemia is a gain-of-function mutation of the PCSK9 gene. The FDA has approved 2 PCSK9 inhibitors (alirocumab and evolocumab) to date, both of which reduce low-density lipoprotein (LDL) cholesterol levels by 50%–60% beyond that what is achieved with statin therapy alone in patients with LDL hypercholesterolemia. Over the past few years, the FDA has expanded its approval of PCSK9 inhibitors to include a larger group of people with elevated LDL cholesterol levels who are at high risk of cardiovascular diseases [34,35]. Amyloid transthyretin (ATTR) amyloidosis is a rare, progressive, and fatal disease characterized by accumulation of amyloid fibrils in organs and tissues. Hereditary ATTR (hATTR) is caused by the mutated TTR gene, which encodes for a protein called transthyretin. Monoclonal antibodies can inhibit the formation of amyloid fibrils deposited in various organs of the body. This clinical trial is underway [36].
4. Antisense oligonucleotide therapy, small interfering RNA, exon skipping therapy
Antisense oligonucleotide (ASO) therapy treats genetic diseases by modulating the expression of specific genes. An ASO is a short chemically modified strand of nucleic acids that can bind to specific regions of RNA. It can be customized for treating extremely rare genetic disease [37]. ASO or small interfering RNA (siRNA) therapy has been approved for treating several rare genetic diseases, including DMD, spinal muscular atrophy, hATTR, and familial hypercholesterolemia. An ASO is used to skip genetic mutations that cause DMD and allow for the production of a truncated yet functional version of the dystrophin protein. The PTC124 is approved for treating DMD with a specific type of mutation and treats patients by skipping exon 53. Many exon skipping therapies for DMD using oligonucleotides are currently in clinical trials [38].
Spinal muscular atrophy (SMA) is a rare genetic disorder caused by mutations in the SMN1 gene, which encodes a survival motor neuron protein. An ASO is used to increase the expression of SMN2, a functional copy of the SMN1 gene,to compensate for the loss of function caused by SMN1 gene mutations. Nusinersen is an ASO that modulates the splicing of SMN2 premessenger RNA to increase the proportion of full-length transcripts, leading to higher levels of functional SMN proteins. It was approved by the FDA in 2016 and Korea. Its price tag was $750,000 for the first year and $375,000 per year thereafter. Another drug for SMA, risdiplam, is an orally administered and centrally and peripherally distributed small molecule (SMN2 splicing modifier) that modulates SMN2 pre-mRNA splicing to increase SMN protein levels. Risdiplam was first approved for use in the United States in 2020 and then was subsequently approved in the EU [39].
Porphyrias are a family of rare diseases mainly caused by inborn errors in heme biosynthesis. Acute episodes of porphyria are caused by the overproduction of heme precursors (hepatic or erythropoietic). The upregulation of 5-δ-aminolevulinic acid synthase 1 (ALAS1) plays a key role in porphyrias. Givosiran, an ALAS1-directed siRNA that was developed to treat acute hepatic porphyria, was first approved in 2019 by the FDA [40]. Vutrisiran and patisiran have recently received marketing authorization for treating hATTR neuropathy. These agents substantially reduce TTR protein levels by degrading TTR mRNA, particularly degrading mRNA via nuclear RNaseH1 using inotersen or the cytoplasmic RNA by forming a silencing complex with patisiran [41]. Nucleic acid drugs of PCSK9 inhibitors are designed to target PCSK9 mRNA and inhibit intracellular protein translation and PCSK9 protein synthesis by occupying or cutting the targeted mRNAby RNase H1. The current PCSK9 nucleic acid inhibitor, inclisiran, developed by Novartis, was administered via subcutaneous injection [34].
Huntington disease is a rare genetic disorder caused by the expansion of a trinucleotide repeat in the HTT gene, which encodes for the huntingtin protein. An ASO is used to target the mRNA encoding the mutant huntingtin protein and reduce its production, which can slow disease progression. Phase III clinical trials are underway [36].
5. Gene replacement and direct genome-editing therapy (Table 3)

FDA/EMA approved gene therapy product for rare monogenic diseases
Gene therapy and genome-editing techniques are promising in treating rare genetic diseases. Gene therapy involves the delivery of functional copies of a missing or defective gene to the cells for correcting the underlying genetic defects. This process, known as gene augmentation, can be achieved using viral vectors, such as adenoviruses or lentiviruses, to deliver therapeutic genes to the appropriate cells. In contrast, gene suppression using siRNA can treat rare diseases caused by the gain of a functional genetic defect [42].
Human gene therapy was first conducted in 1990 in a patient with adenosine deaminase (ADA) deficiency, a rarer and slightly less severe form of severe combined immunodeficiency. However, gene therapy was in the “dark age” owing to the development of adverse events, such as leukemia, or the fatality of vector associated immune reaction. Since the early 21st century, gene therapy has become effective and relatively safe for treating various rare monogenic diseases [40]. Alipogene tiparvovec (Glybera, UniQure, Amsterdam, The Netherlands) is the first gene therapy product approved for lipoprotein lipase deficiency, a rare genetic disorder caused by mutations in the LPL gene. It was approved by the EMA in 2012 but was later withdrawn from the market for commercial reasons. Subsequently, gene therapy products have been increasingly approved for Mendelian disorders by the FDA and the EU. For instance, an autologous CD34+-enriched cell fraction, Strimvelis (GlaxoSmithKline plk, London, UK), which contains CD34+ cells transduced with a retroviral vector that encodes the human ADA cDNA sequence for ADA deficiency (introduced in 2016), and voretigene neparvovec-rzyl (Luxturna, Spark Therapeutics, Philadelphia, USA), for retinal dystrophy caused by mutations in the RPE65 gene, were approved by the FDA in 2017 at a cost of $850,000 per eye [43]. Onasemnogene abeparvovec-xioi (Zolgensma, Novartis, Bazel, Switzerland), another therapeutic for SMA approved by the FDA in 2019, carries a price tag of $2.1 million per treatment [39]. Etranacogene dezaparvovec (Hemgenix, CSL Behring, King of Prussia, PA, USA), an adeno-associated virus vector-based gene therapy for treating adults with hemophilia B (Factor IX deficiency), was approved in 2022, with a price tag of $ 3.5 million, making it among the most expensive drugs worldwide. Betibeglogene autotemcel (Zynteglo, Bluebird Bio, Sommerville, MA, USA) was approved in 2022 to treat β-thalassemia, and hematopoietic stem cells were engineered using a lentivirus to express HBB in patients with certain types of β-thalassemia. Elivaldogene autotemcel (eli-cel; Skysona, Bluebird Bio) was approved in 2022 to slow the progression of neurological dysfunction in boys aged 4–17 years with early active cerebral adrenoleukodystrophy. Skysona (Bluebird Bio)is a single-use treatment that utilizes the patient’s blood stem cells for ex vivo transduction with the lenti-D lentiviral vector to add functional ABCD1 genes to the patient’s hematopoietic stem cells [15].
There are many potentially treatable gene therapies, including lysosomal storage disorders, such as Pompe disease, Gaucher disease, Fabry disease, MPS1, MPS2, and MPS6, although most are currently in phase I/II clinical trial [44]. Genome-editing techniques such as CRISPR-Cas9 and newer-generation genome-editing tools allow for precise and efficient modification of specific regions of the genome [45]. This can be used to correct genetic defects or disrupt the functions of disease-causing genes. Genome-editing can be used to treat a wide range of rare genetic diseases. Examples of diseases potentially treatable with genome-editing technologies include sickle cell anemia, hemophilia, and CF [46]. Currently, there are over 80 phase II and III clinical trials of gene therapy for rare Mendelian disorders [40].
6. Cell therapy
Hematopoietic stem cell transplantation has proven to be effective in many rare genetic disorders. These include Hurler syndrome, Gaucher disease, adrenoleukodystrophy, Canavan disease, severe combined immunodeficiency, Fanconi anemia, and thalassemia. Recently, cell-based gene therapies, such as chimeric antigen receptor T cells, have been engineered to express specific proteins for treating many hematological cancers or lymphomas [15,43]. In addition, liver organ or cell transplantation is promising for treating many inborn errors of metabolism caused by liver-specific enzyme defects such as urea cycle defects and Wilson disease.
7. Drug and target repurposing (Fig. 3)

Process of drug repurposing and sample diseases.
Drug repurposing (also known as drug repositioning or drug reprofiling)is the process of redeveloping a compound for use in different diseases and is now becoming an increasingly important therapeutic strategy for rare diseases. It is based on the scientific principle that drugs often interact with multiple targets or pathways and various drugs may act on the same target or pathway. Compounds tend to exhibit off-target effects that trigger undesired or unexpected adverse events; however, these effects may also be advantageous in other indications. An increasing number of repurposing success stories and companies leveraging repurposing strategies has been reported [47]. Finasteride was originally developed to treat benign prostatic hyperplasia but was eventually repurposed for male-pattern baldness. Another example is liraglutide (Saxenda, Novo Nordisk, Hellerup, Denamark), which was originally developed to treat diabetes mellitus and was repurposed to treat obesity. Drug repurposing is considerably faster and less expensive than traditional drug discovery methods. Specifically, the development risk is reduced as repurposed candidates have already been proven as sufficiently safe in preclinical models and at least in early-stage trials in humans; thus, they are less likely to fail from a safety point of view in subsequent efficacy trials. Moreover, phase I clinical trials of the repurposed drugs can be skipped. The required step that remains is to confirm the efficacy in the new indication at preclinical and clinical levels.
The drug repositioning approach canbe a cost-effective and efficient way to develop therapeutic strategies for rare genetic disorders because drug safety and efficacy have already been established, which can accelerate the development process [48]. According to different estimates, the number of repurposed drugs entering the regulatory approval pipeline is increasing, accounting for approximately 20%–30% of all the drugs approved every year. Some examples of drugs under research for rare genetic disorders using the drug repositioning strategy include rapamycin, initially approved as an immunosuppressant and repurposed to treat tuberous sclerosis complex; losartan, initially approved as an antihypertensive and repurposed to treat Marfan syndrome; doxycycline, initially approved as an antibiotic and repurposed to treat osteogenesis imperfecta; and tyrosine kinase inhibitors used for cancer therapy and repurposed to treat rasopathies such as neurofibromatosis type I and Noonan syndrome [49].
8. mRNA therapy
The mRNA therapy is a relatively new approach to treating diseases that introduces a synthetic mRNA molecule into the body. Once inside the body, mRNA can be translated by the cells into a functional protein. The success of mRNA therapy in overcoming the coronavirus disease 2019 (COVID-19) pandemic has been unfolded. mRNA therapy is an emerging class of therapies, with multiple mRNA-based cancer immunotherapies and vaccines currently in clinical trials in addition to the COVID-19 vaccine. A promising application of mRNA therapy is to restore or augment proteins following its systemic administration, particularly for inborn errors of metabolism such as methylmalonic acidemia caused by methyl malonyl CoA mutase deficiency and PKU caused by phenylalanine hydroxylase defect [50,51]. The advantage of mRNA therapy over viral gene delivery is that the former bypasses the risk of unintended genomic integration, thereby reducing insertional mutagenesis risks. It produces rapid efficacious responses but requires multiple injections because of transient, half-life-dependent protein expression. It is now feasible to systemically deliver lipid nanoparticles targeting hepatocyte cytoplasm. The use of mRNA chemically modified with uridine derivatives enhances translation and reduces immunogenicity, rendering mRNA therapy more suitable for repeated doses. Overall, mRNA therapy has shown great promise for the treatment of rare diseases by providing a targeted approach for delivering missing or defective proteins. Although this approach remains in its early stages of development, it has the potential to revolutionize the treatment of rare diseases and provide new hope to patients and their families.
Process and limitations of clinical trials of orphan drugs for rare genetic disorders
Orphan drugs used to treat rare diseases are generally considered expensive for several reasons. Because rare diseases affect only a small number of people, the market for orphan drugs is limited. Thus, there is less financial incentive for pharmaceutical companies to invest in the development of these drugs and that the costs of research and development must be spread out among a smaller patient population. Developing drugs for rare diseases is more complex and expensive than that for common diseases because the underlying mechanisms of many rare diseases are not well understood and it can be difficult to identify suitable patients for clinical trials. Additionally, the processes of regulatory approval and manufacturing may also be more complex, adding to costs. A lack of competition can lead to higher prices for orphan drugs. Some rare diseases are chronic and require long-term treatment, which can increase the cost of treatment as patients may need to take the drug for many years. Some orphan drugs have unique storage and distribution requirements, which can increase costs. The reimbursement process for orphan drugs can be complex and time-consuming, making it difficult for patients to access these drugs. Additionally, some insurance companies and government healthcare programs may not cover the cost of orphan drugs, making them less accessible to patients [52,53].
There are several challenges to conducting clinical trials on rare genetic disorders [54]:
(1) Patient recruitment: Recruiting sufficient patients with rare genetic disorders to participate in a clinical trial can be difficult, as the patient population is small and the disease may be geographically dispersed.
(2) Genetic heterogeneity: Many rare genetic disorders are caused by different genetic mutations, making it difficult to identify a patient population that is sufficiently homogeneous to participate in clinical trials.
(3) Natural history: The natural history of many rare genetic disorders is not well understood, making it difficult to design appropriate clinical trial endpoints and interpret their results.
(4) Lack of animal models: Many rare genetic disorders lack good animal models, making it difficult to conduct preclinical studies and evaluate the safety and efficacy of potential therapeutics. Although animal models are available, their results are always directly translated to humans.
(5) Limited resources: Conducting clinical trials for rare genetic disorders can be expensive and time-consuming, and many small biotechnological and pharmaceutical companies may not have the resources to conduct them.
(6) Regulatory challenges: The regulatory process for rare genetic disorders can be complex, and it can be difficult to meet the requirements for clinical trial design, data analysis, and submission of regulatory applications.
(7) Ethical concerns: Rare genetic disorders often affect vulnerable populations such as children or individuals with severe life-limiting conditions. This can raise ethical concerns regarding the conduct of clinical trials and the use of experimental therapeutics in these populations.
(8) Reimbursement and access: Even if a drug is approved, the drug access and its reimbursement process can be challenging since most orphan drugs are extremely expensive.
Drug development for rare genetic diseases is a complex and multistep process that typically involves several stages (Fig. 4) [48,55]:

Traditional versus repurposed orphan drug development. PK, pharmacokinetics: PD, pharmacodynamics; IND, investigational new drug; NDA, new drug application.
(1) Discovery and preclinical research: The first step in developing a drug for a rare genetic disease is to identify the geneticmutations that cause the disease and understand the underlying biological mechanisms. This can involve both basic research, such as studying the genetics of a particular disorder, and preclinical research, such as the development of animal models of the disease to test potential therapies.
(2) investigational new drug (IND) application: Once a promising drug candidate has been identified, the next step is to submit an IND application to a regulatory agency, such as the Korean MFDS, the FDA, or the EMA. This application includes information about the drug’s safety, efficacy, and potential side effects as well as details about planned clinical trials.
① Phase I clinical trials: Upon receiving IND application approval, the drug candidate enters a phase I clinical trial. These trials are usually conducted with a small number of healthy volunteers or patients with the target disease, and their main aim is to evaluate the drug’s safety and determine a safe dosage range. Phase 1 clinical trials are often conducted to assess the pharmacodynamics and pharmacokinetics of new drugs.
② Phase II clinical trials: In phase II trials, patients are typically randomly divided into different treatment groups, and the efficacy of the new drug or treatment is compared to a control group or standard of care treatment. Phase II trials may also provide additional information on the pharmacokinetics and pharmacodynamics of new drugs or treatments. These trials are usually conducted in a larger group of patients with the target disease, and their main goal is to evaluate the effectiveness of the new drug or treatment at treating the targeted disease, further assess its safety and tolerability, and identify the optimal dose or dosing regimen.
③ Phase III clinical trials: The purpose of a phase III clinical trial is to confirm the efficacy and safety of a new drug in a larger, diverse population of patients and provide further evidence of its effectiveness compared to existing treatments or a placebo. It ultimately aims to provide sufficient evidence to support the regulatory approval and market launch. Phase III trials may provide additional information regarding the long-term safety and effectiveness of new drugs or treatments.
④ New drug application or marketing authorization application: After the successful completion of phase III clinical trials, the pharmaceutical company will submit new drug applications to regulatory agencies.
Regulatory policies for developing orphan drugs
Orphan drug designation is a regulatory process that provides incentives for the development of treatments for rare genetic disorders. This process provides certain benefits to researchers and drug developers, including tax credits, research and development grants, and exclusive marketing rights once approved. The approval process for orphan drugs can vary among countries but typically involves clinical trials and regulatory reviews to assess treatment safety and. Approximately 15% of the designated orphan drugs are ultimately approved. Once a drug has been approved, it must be authorized for marketing in each country where it is intended to be sold. The process for marketing authorization can vary among countries but generally involves a review of product labeling, manufacturing, and distribution. The duration of exclusive marketing rights is 7 years (United States) or 10 years (Korea, Japan, and the EU)(Table 4) [11,56,57].

Comparison of regulatory policy for the development of orphan drugs
It is important to note that the specific regulations and policies for orphan drugs can vary significantly among countries and can change over time. Orphan drug researchers should be aware of the regulatory landscape in each country where they intend to market their products and work closely with regulatory agencies to ensure compliance.
Conclusion
Orphan drugs have been developed to treat rare diseases, conditions that affect less than 20,000 people in Korea. Owing to the limited patient population, the development of drugs for rare diseases can be challenging for pharmaceutical companies because of development costs and biotechnological issues. To incentivize the development of orphan drugs, governments have implemented various policies and regulations such as tax incentives, research grants, expedited protocol reviews, and market exclusivity periods for orphan drugs. Orphan drug development follows a pathway similar to that of other drugs except that it is tailored to the unique challenges of rare diseases. Overall, orphan drug development is critical for addressing the unmet medical needs of patients with rare diseases.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.