Protective effect of recombinant interleukin-10 on newborn rat lungs exposed to short-term sublethal hyperoxia
Article information

Abstract
Background
Lung injury imposed by hyperoxia is the main cause of bronchopulmonary dysplasia in newborns. These injuries are generated from the early stage of hyperoxia through the main biologic effects of cell death and inflammatory response. Interleukin (IL)-10 is a potent anti-inflammatory cytokine that may have the inhibitory effects on these biologic actions induced by hyperoxia.
Purpose
Based on our former in vitro studies investigating the effect of recombinant IL-10 (rIL-10) on protecting cultured alveolar type II cells exposed to short-term hyperoxia, we performed the in vivo study to investigate the effect of rIL-10 in newborn rats aged P4 exposed to hyperoxia.
Methods
Rats were classified into 3 groups; the control group exposed to normoxia for 24 hours; the hyperoxia group exposed to 65% hyperoxia for 24 hours; and the IL10 group treated with intratracheal instillation of rIL-10 prior to exposure to 65% hyperoxia for 24 hours. Following each treatment, the rats were euthanized. Individual lobes of the right lung were prepared for hematoxyling and eosin (H&E) staining and immunohistochemical staining for thyroid transcription factor-1 (TTF1). Bronchoalveolar lavage (BAL) was performed in the left lung to analyze cell counts and cytokines.
Results
The IL10 group showed preserved air spaces similar to the control group, with decreased cellularity compared to the hyperoxia group, whereas the hyperoxia group showed markedly reduced air spaces with increased cellularity compared to the IL10 group. And, the IL10 group showed more TTF1-positive cells, which represented alveolar type II cells, compared to the hyperoxia group. Inflammatory cells, such as neutrophils and lymphocytes and proinflammatory cytokines of tumor necrosis factor-α, IL-1α, IL-8, and macrophage inflammatory protein-1α were significantly lower in BAL fluid of the IL10 group compared to the hyperoxia group.
Conclusion
These results indicate that rIL-10 may be a promising pharmaceutical measure for protecting newborn lungs from injury induced at the early stage of hyper oxia.
Key message
Lung injury is generated from the early stage of hyperoxia through the biologic effects of cell death and inflammatory response, which eventually leads to evolution of bronchopul-monary dysplasia. Therefore, a protective measure against hyperoxia-induced lung injury is needed. The present study observed that anti-inflammatory cytokine, interleukin-10 had protective effects on newborn rat lungs from injury induced at the early stage of hyperoxia, by preventing cell death and down-regulating inflammatory response.
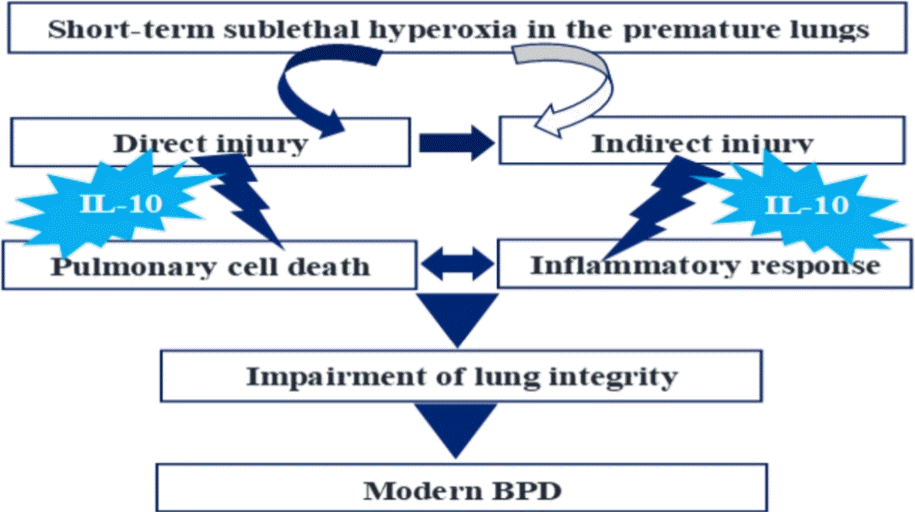
Graphical abstract. BPD, bronchopulmonary dysplasia.
Introduction
Supplemental oxygen is a mainstay in preterm newborns from birth. However, hyperoxia is the most immediate cause of lung injury that eventually leads to evolution of bronchopulmonary dysplasia (BPD).
In recent years, new features of BPD have been recognized as modern BPD, which is characterized by large and simplified alveolar structures and pulmonary vascular malformation with variable interstitial cellularity and/or fibrous hyperplasia [1,2]. of which injuries are accompanied by cytokines released [3] and inflammatory cell influx into the lungs [2]. which eventually might lead to the developmental arrest of the lungs after the first postnatal week [4].
These injurious phenomena including cell or tissue injury and inflammatory reaction begin rapidly following hyperoxia exposure. One study showed that 60% hyperoxia for 2 hours induced histopathological changes with increased inflammatory infiltrates and mitochondrial respiratory chain complexes dysfunction in adult rat lungs [5]. Another showed that 95% hyperoxia for 4 hours induced significant changes in mitochondrial morphology in cultured mouse lung epithelial cells and later, at postnatal day 14 (P14), induced alveolar simplification in mice [6]. Also, our previous study on cultured fetal rat alveolar type II cells (FRATIICs) addressed that 65% hyperoxia induced an increase in cell necrosis after 6–24 hours [7-9] with a decrease in cell proliferation and an increase in the release of IL-8 [8].
Based on these investigations regarding the lung injury during hyperoxia, it is encouraged to pay attention on preventive measures of preterm lungs from injury induced from the early stage of hyperoxia.
In recent years, stem-cell-based therapies have gained concerns as an attractive therapeutic option for treating BPD [10]. However, considering that lung injury begins to be generated within several hours from exposure to hyperoxia [5-9], it may be needed to take a therapeutic approach of lung injury from the perspective of protection before acute injury or sequelae are established, rather than regenerating lung tissues. In addition, it may be a concern that stem and progenitor cells, which are known to be highly sensitive to oxidative stress, could also be another subject to injury from exposure to reactive oxygen species (ROS) produced by hyperoxia, though it was addressed that they are terminally differentiated into the counterparts that are resistant to oxidative damage [11-13]. Therefore, any protective target prior to stem-cell-based therapy may be considered to prevent the lungs from injury induced from the early stage of hyperoxia.
IL-10 is the most potent anti-inflammatory cytokine described to date, and it may have inhibitory effects on hyperoxia-induced biologic actions of cell death and inflammatory response.In recent years, our laboratory has performed systemic in vitro studies on FRATIICs on embryonic day 19 (E19) of gestation to investigate hyperoxia-induced injury and the effect of rIL-10 on the cells exposed to hyperoxia.We found that 65% hyperoxia exposed to FRATIICs induced an imbalance between pro- and anti-inflammatory cytokine, showing upregulation of IL-8 and down-regulation of IL-10 [8], and cellular injury began from the early stage of hyperoxia after 6–36 hours, which was mainly related to cellular necrosis [7-9], and these injuries were alleviated with preincubation of recombinant IL-10 (rIL-10) by decreasing cellular necrosis and IL-8 production, increasing cell proliferation [8]. We also found that IL-10’s signaling proteins of JAK1 and TYK2 suppressed in FRATIICs during hyperoxia were reverted by preincubation of rIL-10 [14].
Based on our former in vitro studies focused on cultured FRATIICs exposed to short-term sublethal hyperoxia, we performed the present in vivo study to investigate the lung injury induced in newborn rat lungs at the early stage of hyperoxia and the pulmonary effects of rIL-10 administered intratracheally prior to exposure to sublethal hyperoxia, under the hypothesis of rapid generation of hyperoxia-induced lung injury in newborn rat lungs and the speculation of protective effects of rIL-10 against lung injury induced at the early stage of hyperoxia.
Methods
1. Animal model
Four-day-old healthy Sprague-Dawley rats were purchased (Daehan Bio, Umseung, Korea). All rat pups were born at term (22 days), weighed 9–9.4 g and were maintained with their dams in the Animal Research Center of Veterinary Medicine throughout the experiments. The rat pups were randomly divided into 3 experimental groups; the control group (C), which was exposed to normoxia for 24 hours; the hyperoxia group (O), which was exposed to 65% hyperoxia for 24 hours; and the IL-10 group (IL10), which treated with intratracheal instillation of rIL-10 prior to exposure to 65% hyperoxia for 24 hours.In the control group (n=10), rat pups were kept with their dams in the standard cage at room air throughout the experiment. Rat pups of the hyperoxia group (n=10) and the IL10 group (n=10) were maintained in the standard cage with their dams and the rat-contained cages were placed in a sealed Plexiglas exposure chamber with continuous monitoring of oxygen concentration at 65% for 24 hours, using an oxygen measuring device, Greizinger GOX 100 Oxygen Meter (Greizinger GmbH, Münzbach, Germany). RIL-10 was reconstituted as a stock solution at 50 μg/mL with sterile phosphate-buffered saline (PBS) added. RIL10 was prepared in an adequate volume of 0.1 mL by adding PBS. Rat pups of the IL10 group were placed in supine position, 4 limbs were restrained, and anesthetized with inhaled ether. A half the tip of the 30-guage needle was prepared bended at a 45° angle. The trachea was exposed by blunt dissection, and a prepared 30-guage needle was introduced into the trachea, and then, rIL-10 (R&D Systems, Minneapolis, MN, USA) was inoculated very slowly at the concentration of 250 ng/g body weight 1 hour prior to exposure to 65% hyperoxia. The skin incision was closed with surgical staples. Nursing dams were rotated every 6 hours between pups in the control and the hyperoxia groups to avoid oxygen toxicity.
After 24 hours, pups were humanely euthanized by an intraperitoneal injection of pentobarbital sodium (60 μg/g body weight). The right lung was excised and then, immersed in 4% paraformaldehyde (PFA) in PBS. The fixed right lung was separated into individual lobes for histology and immunohistochemical staining of TTF1. The left lung was used for BAL to analyze cell counts and cytokines.
2. Bronchoalveolar lavage and cell count
Five-day-old rat pups under the 3 different experimental conditions; C, O, and IL10 group, were placed in supine position, 4 limbs were restrained, and anesthetized with ether. The trachea was exposed by blunt dissection. A 29 G NCV catheter (Forte Grow Medical, Tokyo, Japan) was inserted into the trachea, and 0.2 mL of isotonic saline (kept at 37°C) was gently instilled with a syringe, which was left in the lungs for 5 seconds, and then withdrawn. BAL was performed 3 times. BAL samples obtained for each different group were stored at -80°C. Total cell counts were analyzed from BAL samples on the aliquot fraction with a hemocytometer. Samples were then centrifuged at 150 g for 7 min at 4°C. Cell pellets were resuspended in an adequate volume to obtain 100 cells/mL, and differential cell counts were performed on Cytospin preparations stained with Diff-Quik (Fisher-Scientific, Pittsburg, PA, USA). Manual counts were performed blind by an experienced independent observer to decrease bias. A blinded observer counted a minimum 100 cells to establish the differential count.
3. Concentration of cytokine in BAL fluid
Cytokines in BAL samples stored at-80°C were measured using commercial multiple enzyme-linked immunosorbent assay (ELISA) kits according to the manufacturer’s recommendations (Rat Inflammation ELISA Strip for Profiling Cytok. Signosis, Santa Clara, EA1201; IL-8<GRO/CINC-1>: Assay Designs, Ann Arbor, cat. #900-074).
4. H&E staining for pulmonary tissues
Tissue samples fixed in 4% PFA in PBS were dehydrated through a gradient ethanol series, subjected to hyalinization, and immersed in paraffin. Samples were then embedded in wax and sectioned. The samples were dewaxed using routine procedures, and the tissue slides were stained with H&E; the slides were then observed under the microscope.
5. Immunohistochemistry for TTF1
Tissue samples were processed, sectioned, deparaffinized, quenched in 0.3% H2O2 in methanol, and rehydrated. Slides were then subjected to a 25-minute microwave treatment in a 1 x Antigen Retrieval Citra buffer (BioGenex Laboratories; San Ramon, CA, USA). Immunostaining for TTF1 was enhanced by antigen retrieval, increasing the availability of the antigen. After removal from microwave, slides were allowed to remain in the 1 x Antigen Retrieval Citra buffer for an additional 20 minutes, after which they were rinsed and placed in PBS. Nonspecific staining was blocked by exposing slides for 30 minutes to prediluted normal goat serum (BioGenex Laboratories, San Ramon, CA, USA). The appropriately diluted rabbit anti-TTF1 serum was then applied and incubated for 1–2 hours at room temperature. After washing, a biotinylated goat anti-rabbit immunoglobulin reagent was applied to the slides for 30 minutes (BioGenex SS kit). After washing, a peroxidase-conjugated streptavidin was added and incubated for 30 minutes. To detect peroxidase activity, the sections were subjected to a color reaction with 3,3’-diaminobenzidine tetrahydrochloride containing 3% H2O2 and were lightly counterstained with hematoxylin.
6. Statistical analysis
Results are expressed as the mean±standard devaition. For normally distributed data of cell counts, level of cytokines, and TTF1-positive cells, the intergroup values before and after hyperoxia exposed or before and after rIL-10 treated were analyzed with analysis of variance followed by post hoc tests. P value of <0.05 was considered to be significant.
Results
1. Cell profile in BAL fluid and the effect of rIL-10
Cell profile was analyzed in BAL fluid obtained at postnatal day 5 (P5) for each group of experiments (n=10/group). BAL samples from the hyperoxia group showed a 2.5-fold increase in the proportion of neutrophils compared to the control group (10.3%±2.5% vs. 25.7%±2.1%, P<0.05), whereas the IL10 group showed a 35% decrease in the proportion of neutrophils compared to the hyperoxia group (25.7%±2.1% vs. 16.7%±4.0%, P<0.05) (Fig. 1A). Similarly, cell proportion of lymphocytes from BAL fluid showed a 1.6-fold increase in the hyperoxia group compared to the control group (10.0%±2.6% vs. 15.7%±2.5%, P<0.05), whereas in the IL10 group, showed a 40% decrease compared to the hyperoxia group (15.7%±2.5% vs. 11.7%±1.5%, P<0.05) (Fig. 1A). As shown in Fig. 1A, cell proportion of macrophages showed a 20% decrease in the hyperoxia group compared to the control group (84.7%±4.1% vs. 66.0%±2.6%, P<0.05), and showed a 16% increase in the IL10 group compared to the hyperoxia group (66.0%±2.6% vs. 76.3%±3.2%, P<0.05) (Fig. 1A).

Cell profile in bronchoalveolar lavage (BAL) fluid and the effect of recombinant interleukin-10 (rIL-10). Inflammatory cells in BAL fluid were assessed by hemocytometer counting using Neubauer chamber. (A) Differential cell count is expressed as a percentage of 100 cells observed in high power filed. (B) Total cell count is expressed as a number in BAL fluid. The values are shown mean±standard deviation. C, control group exposed to normoxia for 24 hours; O, hyperoxia group exposed to 65% hyperoxia for 24 hours; IL10, rIL-10-treated group prior to exposure to 65% hyperoxia for 24 hours; BALF, BAL fluid.
Total cell counts increased 1.3-fold in the hyperoxia group compared to the control group (3,433±254 vs. 4,594±324. P<0.05) and decreased 0.85-fold in the IL10 group compared to the hyperoxia group (4,594±324 vs. 3,946±191, P<0.05) (Fig. 1B).
2. Proinflammatory cytokines in BAL fluid and the effect of rIL-10
Proinflammatory cytokines released into the different BAL fluid were analyzed by ELISA for each experimental groups (n=10/group). As shown in Fig. 2, the hyperoxia group showed significant increases in the releases of tumor necrosis factor (TNF)-α, IL-1α, IL-8, and macrophage inflammatory protein-1α (MIP-1α) 2.7-fold, 1.6-fold, 1.8-fold, and 2.6-fold, respectively when compared to the control group (P<0.05) (Fig. 2A–D). And the IL10 group showed significant reductions in the releases of TNF-α, IL-1α, IL-8, and MIP-1α 0.6-fold, 0.85-fold, 0.7-fold, and 0.7-fold, respectively when compared to the hyperoxia group (P<0.05) (Fig. 2A–D). IL-6 showed a little decrease by 4% from BAL fluid in the hyperoxia group compared to the control group (not significant), which did not change significantly in the IL10 group compared to the hyperoxia group (Fig. 2E).
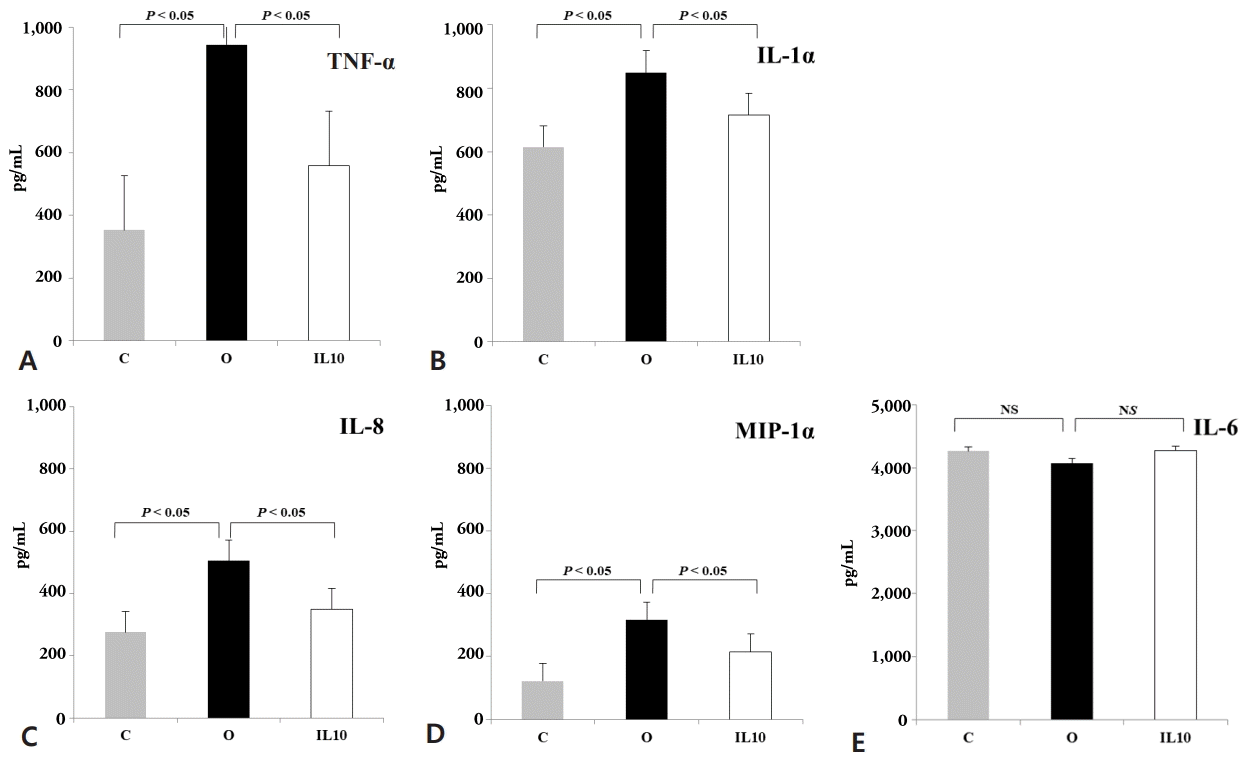
Proinflammatory cytokines and the effect of recombinant interleukin-10 (rIL-10). Proinflammatory cytokines released into the bronchoalveolar lavage fluid were analyzed by enzyme-linked immunosorbent assay. Tumor necrosis factor (TNF)-α (A), interleukin (IL)-1α (B), IL-8 (C), macrophage inflammatory protein-1α (MIP-1α) (D), IL-6 (E). The results are represented as mean±standard deviation. C, control group exposed to normoxia for 24 hours; O, hyperoxia group exposed to 65% hyperoxia for 24 hours; IL10, rIL-10-treated group prior to exposure to 65% hyperoxia for 24 hours.
3. H&E staining of lung tissues and the effect of rIL-10
Representative H&E staining of right middle lung lobe tissue sections for each group are shown in Fig. 3. At 200 magnifications, the lungs of the control group at P5 showed even secondary septa formation with numerous and smaller distal air spaces (Fig. 3A), while the lungs of the hyperoxia group showed a markedly reduced alveolarization, which was represented by fewer, large and irregular air spaces, with increased cellularity (Fig. 3B) compared to the age-matched control lungs (Fig. 3A). The lungs of the IL10 group, however, showed more even alveolar septations (Fig. 3C) with decreased cellularity compared to the lungs of the hyperoxia group (Fig. 3B). At 400 magnifications, the lungs of the control group showed numerous and smaller distal air spaces, which was compatible with normal age-matched lungs (Fig. 3D), while the lungs of the hyperoxia group showed reduced alveolar space with a destructed alveolar septum with increased cellularity (Fig. 3E) compared to the age-matched control lungs (Fig. 3D). However, in the lungs of the IL10 group, alveolar spaces were shown more preserved similar to the control lungs and more even alveolar septa were visible with decreased cellularity compared to the lungs of the hyperoxia group (Fig. 3F).

Conventional histology of lung tissues and the effect of recombinant interleukin-10 (rIL-10). Histological findings (×200 and ×400) were evaluated by hematoxylin/eosin staining of the left lung of newborn age-matched control animals at P5. (A, D) Histology of rat lungs in room air for 24 hours. (B, E) Histology of rat lungs exposed to 65% hyperoxia for 24 hours. (C, F) Histology of rat lungs treated with intratacheal instillation of rIL-10 prior to exposure to 65% hyperoxia for 24 hours. C, control group exposed to normoxia for 24 hours; O, hyperoxia group exposed to 65% hyperoxia for 24 hours; IL10, rIL-10-treated group prior to exposure to 65% hyperoxia for 24 hours.
4. Expression of TTF1 and the effect of rIL-10
Based on the fact that the distribution of TTF1 expression is overlapped with that of surfactant protein A, B, and C [15], we assessed TTF1-expressing cells, as representing alveolar type II cells [15], by immunohistochemical staining on lung sections of each group. As shown in Fig. 4A, the expression of TTF1 was shown lower in the lungs of the hyperoxia group compared to the control group, whereas shown to be increased in the lungs of the IL10 group compared to the hyperoxia group (Fig. 4A). The numbers of TTF1-positive cells decreased 0.82-fold in the hyperoxia group compared to the control group (513±70 vs. 419±73, P<0.05) and increased 1.2-fold in the IL10 group compared to the hyperoxia group (419±73 vs. 509±22, P<0.05) (Fig. 4B).

Alveolar type II cells by immunohistochemical stains of thyroid transcription factor-1 (TTF1) and the effect of recombinant interleukin-10 (rIL-10). (A) Distribution of alveolar type II cells (×200) was evaluated by assessing TTF1-expressing cells by immunohistochemical staining on lung sections of age-matched control animals at P5. TTF1-expressing cells in lung tissue are shown as brown. (B) TTF1-positive cells are expressed as number. Values are shown as mean±standard deviation. C, control group exposed to normoxia for 24 hours; O, hyperoxia group exposed to 65% hyperoxia for 24 hours; IL10, rIL-10-treated group prior to exposure to 65% hyperoxia for 24 hours.
Discussion
The main findings of the present study are that exposure to 65% hyperoxia for 24 hours led to pulmonary tissue impairment with diminished distribution of alveolar type II cells in the lungs and a significant increase in proinflammatory cytokines and influx of inflammatory cells, and that these injurious processes induced at the early stage of hyperoxia were prevented by exogenous IL-10 instillation prior to exposure to hyperoxia.
We conducted these experiments on newborn rats aged P4 and 5, which is based on the facts that P4-5 of rats corresponds to late saccular and early alveolar stage for the rat lung development, which is important stage when classical alveolarization begins [16]. We conducted this study using newborn rats, which is based on the fact that the morphologic changes of BPD resemble hyperoxic lung injury in newborn rodents [17-19].
Exposure to hyperoxia causes direct oxidative cell damage through the production of ROS [20] and a secondary inflammatory response in the lungs [21]. All of these pathologic alterations converge toward the central event, alveolar cell death [20].
Alveolar type II cells are key components of the alveolar structure. Not only do they secret surfactant, but they also restore the alveolar epithelium after acute lung injury [22] and function as mechanosensory cells in response to stimuli such as hyperoxia [23]. Also it had been indicated that failure of type II cells to proliferate during the first week of life may permanently alter postnatal lung development during a critical period of postnatal lung development [4]. In the present study, we examined alveolar type II cells by evaluating TTF1 expression. TTF1 is a critical determinant of lung epithelium-specific gene expression and lung morphogenesis [24,25]. TTF1 activates the expression of surfactant protein A, B, and C gene transcription. Hence, the location of expression of TTF1 is consistent with distribution of surfactant protein A, B, and C in alveoli [15]. TTF1 is expressed in the rat fetal lungs from the earliest period of lung bud formation [26].In human lungs, with advancing development, TTF1 expression is not detected in type I cells, and between 36–42 weeks of gestation, type II cell nuclei are intensely labeled by anti-TTF1, and after birth, it is stably expressed in alveolar type II cells [27].
In the present study, we observed that TTF1 expression was lower in the hyperoxia group than the control group. These findings are supported by our previous studies showing that alveolar type II cells cultured from rats at E19 generally showed necrotic aspect during the acute stage of hyperoxia at 6–36 hours rather than apoptosis, with inhibition of cell proliferation [7-9], and with activation of proteolytic enzyme, cathepsin B [9], which contributes to cell death as a major non-caspase protease released by lysosomal permeabilization [28,29] that is induced by oxygen radical species (ORS) [29].
These cellular injuries including cell death should lead to the release of proinflammatory mediators, damaging neighbor cells. Therefore, a reduction in the alveolar type II cell population would likely affect lung integrity and repair.
In the present study, we found that 65% hyperoxia for 24 hours induced a markedly reduced alveolarization represented by fewer, large and irregular air spaces with destructed alveolar septum and increased cellularity, of which findings were consistent with the pathologic features of modern BPD [1,2], while the control lungs showed even secondary septa formation with numerous, smaller and evenly distributed distal air spaces, which was compatible with normal P5 age-matched lungs. In rat lung development, P5 corresponds to the early alveolar stage when the alveolar formation begins [16]. These results were consistent with other investigations showing alveolar deformation in rodents starting in the acute stage of exposure to hyperoxia; from 4 hours of exposure to 95% hyperoxia to neonatal mice <12 hours old [6]; from 2 hours of exposure to 60% hyperoxia to adult rats [3]; and from 90 minutes of exposure to 75% hyperoxia to adult rats [30].
All of the cell injury processes induced during hyperoxia are connected to the release of proinflammatory cytokines, including preformed proinflammatory cytokines in pulmonary cells.
In the present study, the chemokines IL-8 (CINC-1) and MIP-1α and the proinflammatory cytokines of TNF-α and IL-1α were significantly upregulated within 24 hours of 65% hyperoxia. The present results about the upregulation of chemokines and cytokines were consistent with those of other studies on newborn rats that showed a rapid increase in chemokines such as IL-8 and MIP-2 and cytokines including TNF-α, IL-6, IL-1α, and IL-1β immediately after exposure to hyperoxia [31-33]. Preformed and released chemokines and cytokines play a pivotal role in the recruitment of nucleated cells from the plasma into the lungs [31,32], and recruited nucleated cells release additional cytokines and oxygen radicals in the lungs. In conformity with these backgrounds,the present study showed a significant increase in neutrophils from BAL fluid of the hyperoxia group compared to the control group. The present results were similar to those of a study on the BAL fluid of 6-day-old rats exposed to 65% hyperoxia for 24 hours [34].
Similarly, the present study showed a significant increase in lymphocytes in BAL fluid of the hyperoxia group compared to the control group, which may be explained by the fact that innate and adaptive immunity act in union, therefore cytokines released from cells of the innate immune system can prime lymphocytes [35]. These primed lymphocytes have immune response following exposure to a specific stimulus such as hyperoxia [36].
Unlike with neutrophils and lymphocytes, the present study showed a decrease in the percentage of macrophages in BAL fluid from the hyperoxia group compared to the control group. Our results regarding macrophages could be supported by the previous studies showing that exposure to 95% hyperoxia for 24 hours induced alveolar macrophage growth arrest with limited death, including morphological changes in cultured macrophage cells [37], 65% hyperoxia for 7 days decreased the expression of macrophages in the lung tissues of newborn mice [38]. Additionally, according to a previous report, alveolar macrophages were first identifiable at postnatal day 1 in mice and peaked in numbers at P5 [39]. Our finding of decreased macrophages in the BAL fluid of the hyperoxia group at P5 may imply that sublethal hyperoxia may preferentially induce the inhibition of macrophage proliferation or their limited cell death at the very early stage of hyperoxia.
Collectively, our results about inflammatory cells in BAL fluid may imply that the inflammatory response starts in the very early stage of hyperoxia exposure in newborn rat lungs, where neutrophils may act very rapidly as the first immune cells to be recruited during short-term sublethal hyperoxia.
Given these injurious processes induced during hyperoxia, anti-inflammatory strategy could be promising to protect the lungs from injury that begins from the very early stage of hyperoxia.
IL-10 is widely known as a potent anti-inflammatory cytokine, and its effects on protecting lungs or pulmonary cells had been defined in adult rodent lungs or FRATIICs under the conditions of inflammation [40], mechanical ventilation [41], mechanical stretching [42], or hyperoxia [8,43],
In recent years, our laboratory investigated the protective effect of IL-10 on cultured FRATIICs exposed to 65% hyperoxia for 6–36 hours [7-9]. We found that in FRATIICs during 65% hyperoxia,IL-10 treatment reduced alveolar type II cell necrosis and the release of IL-8, and inhibited the activation of the proteolytic enzyme, cathepsin-B [9] and reverted the suppressed expression of IL-10 signaling proteins [14]. Based on our in vitro studies on alveolar type II cells, we conducted the present in vivo study on newborn rats, in which we gave rIL-10 intratracheally prior to exposure to hyperoxia. We used a dose of 250 ng/g body weight of rIL-10, based on the previous study showing that intratracheal instillation of rIL-10 at a concentration of 250 μg/kg body weight had effects on attenuating pulmonary inflammation in adult rats [44].
The present study showed that rIL-10 instillation prevented the loss of alveolar type II cells and impairment of pulmonary architecture, inducing down-regulation of proinflammatory cytokines and reducing the influx of neutrophils in neonatal rats during the early stage of hyperoxia. The mechanisms by which IL-10 protects lungs injured by short-term sublethal hyperoxia are not fully defined. However, some plausible explanations can be considered regarding the present observations.
The first explanation is that IL-10’s direct effect on inhibiting nuclear factor kappa B (NF-κB) activation may alleviate direct cell injury and inflammatory response induced by hyperoxia. NF-κB activation is an indicator of oxidative stress [45,46], and, as a pivotal transcription factor modulating inflammation, is implicated in mediating the release of proinflammatory cytokines under hyperoxia [47] before increases in proinflammatory cytokines are detected [48]. Such NF-κB activation increases in rat alveolar type II cells in response to sublethal hyperoxia [49]. and there have been many reports showing IL-10’s anti-inflammatory effect on inhibiting NF-κB activity [50,51]. In addition, IL-10’s capacity to protect pulmonary cells from direct injury by inhibiting NF-κB activation may be related to its ability to suppress phospholipase A2 expression secondary to low doses of oxygen [52] and to inhibit cathepsin B activation [9]. Low doses of ORS activate phospholipase A2 first, consequently inducing lysosomal rupture indirectly through the destabilization of lysosomes [52], in which activation of phospholipase A2 is inhibited by the NF-κB-inhibitor [53]. In terms of cathepsin B-mediated lung injuries, cathepsin B-mediated alveolar macrophage death was defined in generating lung injures [54], and cathepsin B-mediated human or rat epithelial cell death was defined with stimulation of lipopolysaccharides [29] or hyperoxia [9]. In addition, IL-10 has been shown to inhibit such cathespin-B activation in FRATIICs exposed to hyperoxia [9] and in tumor cells including lung cancer cells [55].
The second explanation is that IL-10 may affect ORS-induced direct cell damage by down-regulating the expression of inducible nitric oxide synthase and reducing nitric oxide (NO) that is excessively released in lungs exposed to hyperoxia [43,56]. Excessive NO is directly cytotoxic and injure cells and tissues, and can cause cell death, through reacting with the primary oxygen radical, superoxide anion [43,56].
The third explanation is that IL-10 may indirectly influence IL-1 or TNF-α production by suppressing their receptors. This explanation is supported by a study showing that TNF-α receptor II and IL-1 receptors I/II increased significantly in IL-10 knock-out mice when compared to wild-type mice following lipopolysaccharide-stimulated lung injury [57].
The fourth explanation is that all these effects of IL-10 may prevent or inhibit the influx of inflammatory cells into the lungs and could prevent the additional injury from the cells recruited.
The present study was the first in vivo study to investigate lung injury generated at the early stage of hyperoxia in newborn rats and the effect of the anti-inflammatory cytokine, IL-10 instilled intratracheally to newborn rat lungs prior to exposure to short-term sublethal hyperoxia. However, the present study would be a preliminary experiment as the first in vivo study to figure out general profile of lung injury and rIL-10 effects on newborn rat lungs exposed to short-term sublethal hyperoxia.It is required to be more explored to understand IL-10’s effects on lung tissue exposed to hyperoxia through variable rodent lung models and with more specimens.
In summary, our in vivo study on newborn rats showed that rIL-10 instillation inhibited the loss of alveolar type II cells and prevented impairment of pulmonary structures, thereby reducing the release of proinflammatory mediators and the influx of inflammatory cells into the lungs during the early stage of hyperoxia. In generating lung injury, cell death and inflammation reciprocally amplify each other [58]. Our investigations indicate that IL-10 is a promising pharmaceutical measure for protecting neonatal lungs from injury during the early stage of sublethal hyperoxia by preventing pulmonary cell death and inflammatory response.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author contribution
Conceptualization: HSL; Data curation: HSL; Formal analysis: HSL; Methodology: HSL, YJR, MJL; Project administration: HSL; Visualization: HSL; Writing-original draft: HSL; Writing-review & editing: HSL
Acknowledgements
I thank Prof. Ryu, Young-Joon and Lee, Min-Jae for their technical support in carrying out the present research.