Peripheral nerve sheath tumors in the head and neck in patients with APC gene deletion mutations: a case report and scoping review of the literature
Article information

Abstract
Adenomatous polyposis coli (APC) is a tumor suppressor gene expressed throughout the body. APC mutations increase the risk of malignancy and are often characterized by syndromes that encompass a spectrum of neoplastic manifestations, such as familial adenomatous polyposis (FAP). We present a rare case of palatal peripheral nerve sheath tumor in the context of APC gene mutation. A 17-year-old male with a significant history of FAP presented to our clinic with globus sensation for 5 months with increasing discomfort. Flexible nasolaryngoscopy revealed a pedunculated lesion attached to the posterior surface of the soft palate. Imaging was obtained and confirmed a soft tissue homogenous mass contiguous with the soft palate. Endoscopic-assisted transoral resection was performed and pathologic features were consistent with schwannoma. We also discuss the spectrum of benign neoplastic lesions. Current literature fails to describe pharyngeal masses in the setting of APC gene mutations. The purpose of this case report is to describe a patient presentation of a symptomatic pharyngeal tumor with a known APC gene mutation and explore the differential diagnoses that must be considered.
Key message
In this report, we describe our experience with a patient with an APC-related genetic syndrome who presented with a rare palatal lesion with characteristics of a schwannoma. We discuss the role of immunohistochemical staining in discerning the differential diagnosis.
Introduction
The adenomatous polyposis coli (APC) gene is a tumor suppressor gene on chromosome 5q21 that is expressed throughout the body and encodes a 312 kDa protein [1-3]. Its ability to act as a tumor suppressor relies on its interactions in the Wnt/beta-catenin signaling pathway. Functional APC proteins form a complex with beta-catenin resulting in phosphorylation, ubiquitination, and subsequent destruction of beta-catenin. In the presence of Wnt, beta-catenin is freed from the destruction complex and translocates to the nucleus to activate transcription factors [4,5]. Without a functional APC protein, beta-catenin is constitutively active resulting in abnormal gene transcription and neoplastic lesions.
APC-associated gene mutations may arise de novo or be inherited in an autosomal dominant pattern resulting in a variety of neoplastic growths [6]. Familial adenomatosis polyposis (FAP) is an autosomal dominant disorder notable for numerous APC-associated adenomatous polyps within the gastrointestinal tract. It is often described as a spectrum of syndromes that include Gardner and Turcot [7]. Gardner syndrome presents with FAP-associated intestinal polyps, osteomas, and desmoid tumors [8], while Turcot syndrome presents with intestinal polyps and central nervous system tumors [9]. Additional FAP-associated growths have been identified in the liver, abdominal wall, and odontomaxillary spaces [10,11].
APC-related manifestations presenting in the nasopharynx and neck are not well described, likely due to their rarity. The differential includes benign and malignant peripheral nerve sheath tumors that are promoted through the activation of Wnt signaling [12]. Nasopharyngeal angiofibromas and thyroid carcinomas have been reported as extracolonic manifestations of FAP [10,13-15]. Therefore, pharyngeal lesions in the setting of an APC-related genetic syndrome warrant further exploration. In this report, we describe our experience with a patient who presented with a rare palatal lesion and we discuss the role of immunohistochemical staining in discerning the diagnosis.
Case presentation
A 17-year-old male known for an APC 2434del4 mutation, colonic polyposis, a mandible osteoma, and poststreptococcal reactive arthritis presented to the Ohio State University Wexner Medical Center Otolaryngology clinic due to a globus sensation with increasing discomfort. Flexible nasolaryngoscopy showed a nasopharyngeal mass. Lab results at the time of presentation included elevated C-reactive protein and normal complement and lactate dehydrogenase levels. Computed tomography (CT) of the neck on August 7, 2023 demonstrated a 2.3×2.1×1.3-cm midline mass in the posterior pharynx contiguous with the soft palate (Fig. 1). Lung CT additionally showed four lower lobe 2–3 mm nodules bilaterally (left lower lobe: 3 mm, 3 mm, and 2 mm and right lower lobe: 3 mm).

Computed tomography neck demonstrated a midline mass (arrow) contiguous with the soft palate in the pharynx.
Due to symptomatic manifestations and concern for malignancy, the patient underwent elective surgical removal of the mass. Intranasal and oral approaches were utilized. The mass was tan-yellow in color, friable, and bled upon manipulation (Fig. 2). Following removal, the tumor was sent to pathology for further characterization.

The neoplasm was appreciated intraoperatively from the oral cavity (A) and nasopharynx (B) on flexible laryngoscopy. (C) The area was reassessed in the operating room after the lesion was removed from the nasopharynx.
Pathology identified the mass as a 2.7×1.8×1.6-cm benign peripheral nerve sheath tumor that predominately showed features of a schwannoma (Fig. 3). The lobulated and nodular soft tissue mass was tan-pink to yellow in color and included a thin, mucosa-like fibromembranous covering and focal hemorrhagic material. Histology demonstrated hypocellular (Antoni B) and hypercellular (Antoni A) areas of ovoid cells in a myxoid to hyalinized stroma with a focal nuclear palisading pattern. No clear mitotic figures were appreciated. Mild degenerative cytologic atypia and cystic degeneration was present. Regions of necrosis showed ulceration of overlying squamous mucosa. Immunohistochemistry stains were diffusely positive for S-100 and calretinin. Peripheral staining was positive for CD34, glucose transporter protein 1 (Glut-1), and epithelial membrane antigen (EMA). The tumor was negative for Desmin, smooth muscle actin, and nuclear beta-catenin.

(A) Variable hypocellular and hypercellular areas with focally hyalinized stroma (H&E, ×10). (B) Verocay bodies with nuclear palisading (H&E, ×20). (C) S100 immunohistochemical stain with diffuse nuclear and cytoplasmic positivity (S100, ×20).
Throughout the surgical case, pathologic assessment, and write-up, appropriate patient information was protected and completely de-identified.
Differential diagnoses of nasopharyngeal masses
A variety of benign and malignant differential diagnoses were considered which included neurofibroma, neuroma, malignant peripheral sheath tumor, granular cell tumor, neural sheath myxoma, and Juvenile nasopharyngeal angiofibroma (JIA) (Table 1) [16-37]. Our patient’s mass was positive for S-100 indicating a peripheral nerve sheath tumor originating from Schwann cells [38]. However, schwannomas, neurofibromas, neuromas, malignant peripheral nerve sheath tumors, granular cell tumors, and neural sheath myxomas all demonstrate positive S-100 staining due to their cellular origination. Additional staining patterns and microscopic features were necessary to further differentiate which type of peripheral nerve sheath tumor was present.
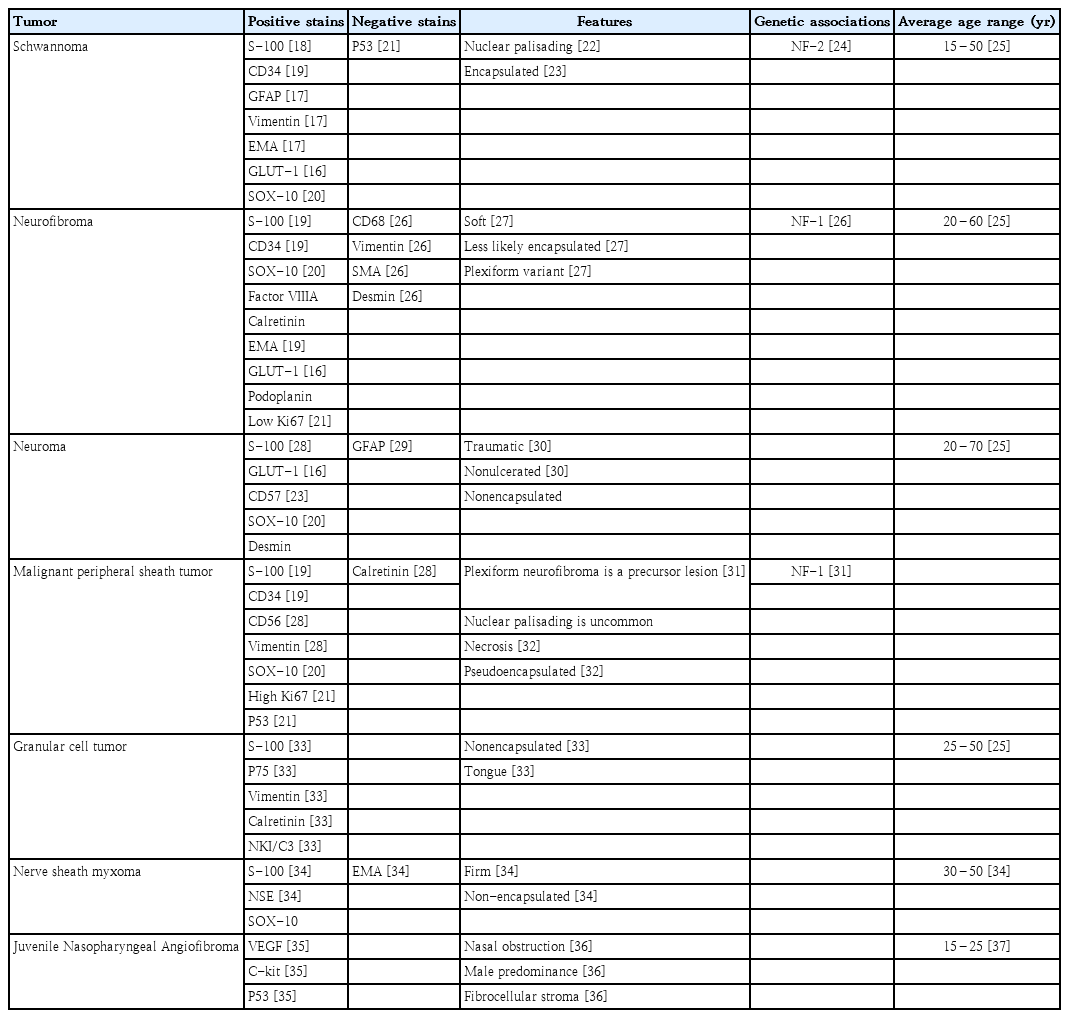
Notably, our patient’s mass was also positive for GLUT-1 and EMA indicating encapsulation, seen as a thin rim of tissue demarcating the mass [16,17]. Of the masses on our differential, schwannomas and neurofibromas are most likely to be encapsulated. Although neuromas, granular cell tumors, and nerve sheath myxomas stain positively for S-100, neither are typically encapsulated as seen in this patient.
Moreover, malignant peripheral sheath tumors stain positively for S-100 and CD34 and demonstrate areas of necrosis as in our patient and often present with pseudoencapsulation; however, patterns of nuclear palisading are extremely uncommon. JIA is a common diagnosis of nasopharyngeal polyps in a younger patient population without a history of neoplastic growths. Yet, immunohistochemistry staining patterns and clinical features in our patient did not support the diagnosis of JIA.
Taken together, neurofibromas and schwannomas share a similar positive and negative staining profile to the mass removed from our patient and were highest on the differential diagnoses. However, patient demographics, histologic findings of encapsulation, and Antoni A and B bodies favor the diagnosis of a schwannoma (Table 2) [23-26,39,40].

Discussion
This 17-year-old patient had a familial APC 2434del4 mutation and a history of colonic polyposis and a mandible osteoma, known manifestations of APC-related gene mutations. The nasopharyngeal mass excised from our patient with Gardner syndrome demonstrated multiple characteristics of a schwannoma, a rapidly growing benign peripheral nerve tumor originating from Schwann cells. Macroscopic excision demonstrated a clearly defined, exophytic mass with mucosal ulceration.
We additionally sought to broadly describe the immunohistochemical and histological features of nasopharyngeal neural tumors. Schwannomas often masquerade as other nerve sheath tumors and must be differentiated through staining patterns and microscopic features as demonstrated by this patient [18]. Distinctive positive immunohistochemical staining for S-100, CD34, Vimentin, GLUT-1, and EMA are often seen. Additionally, histologic growth patterns of Antoni A, described as hypercellular, nuclear palisading bodies, and Antoni B patterns, characterized as hypocellular arrangements accompanied by myxoid stroma support a schwannoma diagnosis.
Schwannomas may arise from anywhere along the peripheral nervous system. Rare locations of schwannoma presentation include the lungs [41], colon [42], and nasopharynx. Such diagnoses rely on immunohistochemical staining, commonly S-100 staining, and histopathologic examination, as seen in our patient. Notably, incidental lung masses were found on CT imaging our patient during the nasopharyngeal mass workup. The possibility of additional schwannomas present in this patient cannot be ignored and further workup is recommended.
Patients with nasopharyngeal schwannomas often present with long-standing complaints of nasal obstruction, snoring, and a globus sensation [43]. Rare symptoms include coughing, dysphagia, and even hearing loss [44]. Preoperative biopsy is not routinely performed due to its vascular nature in the nasopharynx. Proper operative planning including CT and magnetic resonance imaging can provide anatomic localization. Gold standard therapy is total excision resulting in a low risk of locoregional recurrence [45].
Recent advancements in genetic testing of tumors have allowed for identification of the entire genetic profiles. For instance, NF1, NF2, and CDKN2C are some of the most frequently mutated genes in spinal schwannomas [46]. Few cases of APC mutations manifesting into a schwannoma are known, and presentation of a nasopharyngeal schwannoma in the setting of FAP specifically has not been previously reported. One case report has identified a loss of APC within a cranial schwannoma via intragenetic markers [47]. However, the exact mechanism of APC mutations leading to schwannomas remains unclear. APC has shown a role in regulating axonal development and myelination in the peripheral nervous system [48]. A disruption in this mechanism may lead to the unregulated schwannoma growth. Further research is required to better understand the genetic landscape of schwannomas.
Postoperative follow-up studies for pharyngeal masses in the background of an APC mutation has not been established. However, the overall prognosis is optimistic. We recommend prompt removal of symptomatic pharyngeal masses due to the high risk of neoplastic transformation and to prevent airway obstruction. After complete resection of benign tumors, radiation and chemotherapy may be omitted [49]. In addition, typical surveillance and prophylactic procedures for patients with APC mutations should be continued.
In conclusion, schwannoma should be considered in patients with an exophytic nasopharyngeal mass. This case report highlights the differential diagnosis of a nasopharyngeal polyp and describes the novel presentation of a pharyngeal tumor demonstrating schwannoma features in the setting of a familial APC gene mutation.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author Contribution
Conceptualization: MA, TC; Data curation: MA, MC; Formal analysis: KMB, MA, TC; Methodology: KMB, MA; Project administration: TC; Visualization: KMB, MC; Writing-original draft: KMB, MA; Writing - review & editing: KMB, MA, TChiang