Multiomics approaches in Kawasaki disease: insights into pathogenesis and emerging directions for diagnosis and treatment
Article information

Abstract
Kawasaki disease (KD) is an acute febrile vasculitis and the leading cause of acquired heart disease in children. Despite decades of research, the etiology remains unknown and key mechanisms linking systemic inflammation to coronary artery lesions are incompletely defined. High-throughput technologies—including genomics, transcriptomics, proteomics, metabolomics, epigenomics, and immunomics—have enabled systems-level profiling of KD and highlighted reproducible inflammatory and vascular pathways. Multiomics integration increasingly supports convergent mechanistic axes, particularly interleukin (IL-1/IL-6–neutrophil programs, Fcγ-receptor signaling related to intravenous immunoglobulin (IVIG) pharmacodynamics, Ca²+/nuclear factor of activated T cells-dependent T-cell activation, and endothelial/extracellular matrix remodeling associated with coronary outcomes. While these findings provide a robust framework for biomarker discovery and therapeutic hypothesis generation, most signatures remain investigational and require prospective validation, standardized sampling (pre-/post-IVIG), and clinically scalable assays before routine implementation. This review summarizes current multiomics applications in KD, prioritizes the most consistently supported pathways, and outlines a pragmatic roadmap toward clinically useful risk stratification, disease monitoring, and outcome prediction.
Introduction
Kawasaki disease (KD) is an acute, self-limited systemic vasculitis that predominantly affects children under 5 years of age. Despite timely intravenous immunoglobulin (IVIG) therapy significantly reduces the risk of coronary artery lesions (CALs), the underlying etiology and molecular mechanisms remain incompletely understood. Traditional immunological and clinical studies have provided important insights into KD pathogenesis; however, they often fail to capture the disease’s multi-layered molecular complexity spanning genetic, immunologic, metabolic, and environmental dimensions.
The advent of high-throughput multiomics technologies— including genomics, transcriptomics, proteomics, metabolomics, and epigenomics—has enabled comprehensive, systems-level analyses that link molecular alterations to immune dysregulation and vascular injury. By integrating diverse omics layers, researchers can now trace the biological cascade from genetic susceptibility to immune activation and endothelial dysfunction. This holistic approach offers a powerful framework for identifying biomarkers and redefining disease subtypes in KD. At present, however, most multiomics findings primarily provide mechanistic and hypothesis-generating insights, serving as a foundation for future translational studies rather than enabling immediate clinical implementation.
Overview of multiomics technologies in KD
1. Genomics
1) Genetic susceptibility and population specificity
Genomic investigations have begun to refine the concept of KD as an idiopathic disease with underlying polygenic susceptibility, in which host genetic background interacts with environmental triggers. Although the precise cause remains unknown, susceptibility patterns—such as higher incidence in East Asian populations and familial aggregation— suggest contributions from population-enriched alleles alongside shared environmental exposures [1].
Genome-wide association studies (GWAS) have identified reproducible susceptibility loci converging on immune and vascular pathways. The earliest and most robust finding involves inositol 1,4,5-trisphosphate 3-kinase C (ITPKC), where a functional single nucleotide polymorphism (rs28493229) alters splicing efficiency and the Ca²+/nuclear factor of activated T cells (Ca²+/NFAT) signaling pathway, augmenting T-cell activation and inflammasome/IL-1 responses, thereby conferring increased aneurysm risk [2-4]. Variants in Fc gamma receptor IIa (FCGR2A) (rs1801274) implicate Fcγ-mediated IgG interactions in KD pathogenesis and IVIG pharmacodynamics. However, large-scale studies indicate that while FCGR2A/2B/3 polymorphisms influence disease susceptibility, they do not consistently predict IVIG resistance or coronary outcomes [5-8)].
Additional loci—including caspase 3 (CASP3) (affecting NFAT binding and apoptosis regulation) [9-11], B lymphoid tyrosine kinase (BLK), CD40, human leukocyte antigen (HLA) class II genes, and vascular remodeling factors such as matrix metalloproteinase (MMP) haplotypes, angiopoietin-1 (ANGPT1), and vasoactive endothelial growth factor A (VEGFA)—underscore the interplay between adaptive immunity and endothelial integrity [1,12-14]. Genetic variations in chemokine and transporter genes (CC chemokine receptor 5 [CCR5], CC chemokine ligand 3-like 1 [CCL3L1], ATP-binding cassette subfamily C member 4 [ABCC4]) further supports the involvement of leukocyte trafficking and vascular homeostasis [15,16]. Collectively, these loci define overlapping pathogenic axes encompassing immune hyperreactivity (Ca²+/NFAT, Fcγ, IL-1), adaptive immunity (B/T-cell activation, HLA), and vascular stability.
2) Rare variants and pharmacogenomics
Rare-variant analyses from whole-exome and whole-genome sequencing (WES/WGS) extend these insights by identifying alleles enriched in patients with severe vascular phenotypes, particularly CAL. Variants in nebulette sarcomeric isoform (NEBL), tubulin, alpha 3c (TUBA3C), tumor necrosis factor receptor-associated factor 5 (TRAF5), and MAM domain containing GPI anchor 1/2 (MDGA1/2)—genes linked to vascular or cytoskeletal structure—suggest that determinants of aneurysm formation may be distinct from those driving overall disease susceptibility [17].
Pharmacogenomic WGS studies in multiancestry cohorts have also revealed candidate variants associated with IVIG nonresponsiveness, such as fibronectin type III and ankyrin repeat domains 1 (FANK1), mitogen-activated protein kinase kinase 3:potassium inwardly rectifying channel subfamily J member 12 (MAP2K3:KCNJ12), mannosidase alpha class 1A member 2 (MAN1A2), endothelin-1 (EDN1), and Scm-like with 4 MBT domains 2 (SFMBT2), underscoring the polygenic and ancestry-dependent nature of treatment outcomes [18].
3) Population and ancestry-specific effects
Population specificity remains a critical theme. Many initial discoveries arose from East Asian cohorts, where KD incidence is highest, with loci such as ITPKC showing stronger effect sizes in these populations [3]. Others, including FCGR2A and HLA class II, replicate across ancestries but with variable effect sizes or allele frequencies [8,13]. Recent European and multi-ethnic GWAS/WES efforts have confirmed core KD susceptibility loci (such as ITPKC and FCGR2A) and in some cases uncovered ancestry-specific risk variants, with many of these genes implicated in immune regulation [2,8,10,19]. These findings emphasize the need for larger, ethnically diverse datasets and fine-mapping that account for local linkage disequilibrium and allele frequency differences.
4) Translational implications
From a translational standpoint, genomics provides new opportunities for precision medicine in KD. Genotype-informed risk stratification may complement existing clinical scoring systems (e.g., Kobayashi, Egami, Sano), which demonstrate high predictive accuracy in Japanese cohorts but perform less effectively in North American populations [20,21]. Pharmacogenomic markers hold promise for identifying IVIG nonresponders, though single-gene predictors remain insufficient. Integrative approaches combining genomic, transcriptomic, and proteomic features are more likely to yield clinically actionable models.
Notably, convergent genetic evidence implicating the Ca²+/NFAT and IL-1 signaling pathways—especially via ITPKC and related gene variants—supports the rationale for targeted adjunctive therapies such as calcineurin inhibitors or IL-1 blockade in genetically defined high-risk subgroups [22,23].
2. Transcriptomics
1) Bulk transcriptomic insights into innate immune activation
Transcriptomic profiling has substantially deepened our understanding of KD immunopathogenesis. Bulk RNA-seq analyses consistently reveal a dominant innate immune signature during the acute phase, characterized by marked up-regulation of neutrophil-associated genes and IL-1/IL-6 signaling pathways, with interferon-stimulated genes variably enriched across cohorts [24-26]. Modules related to neutrophil degranulation, protease activity, and S100 family members robustly distinguish KD from other febrile illnesses and correlate with systemic inflammation and CAL risk [27,28]. Meta-analyses of bulk RNA-seq across human and murine datasets confirm that acute KD is driven by innate immune activation, neutrophil effector programs, and IL-1/IL-6 signaling enrichment [29].
2) Single-cell RNA sequencing reveals immune heterogeneity
Recent advances in single-cell RNA sequencing (scRNA-seq) have refined the observations from bulk RNA-seq analyses by resolving cellular heterogeneity in KD. Early scRNA-seq studies on peripheral blood demonstrated excessive neutrophil activation and innate immune dysregulation compared with controls [30], while analyses of IVIG-resistant patients revealed immune shifts associated with CAL formation [31]. A landmark study in human peripheral blood mononuclear cell (PBMC) further showed that monocytes display hyperinflammatory signatures with reduced antigen-presentation capacity during the acute phase, suggesting coordinated crosstalk among monocytes, neutrophils, and T cells [32].
Integration of scRNA-seq and bulk RNA-seq further revealed alterations in T, B, and natural killer cell compartments, including shifts in T-cell differentiation trajectories toward regulatory or alternative states and aberrant activation of mTOR and infection-response pathways in multiple immune subsets [33]. In CAL patients, scRNA-seq profiling of over 200,000 PBMCs identified distinct inflammatory immune subpopulations differentiating CAL from non-CAL cases, suggesting that peripheral immune states mirror coronary vascular injury predisposition [34].
3) Meta-analytic convergence and shared neutrophil programs
A recent large-scale, multicohort single-cell meta-analysis integrating over 500,000 immune transcriptomes from KD, multisystem inflammatory syndrome in children (MIS-C), and healthy controls identified a conserved CD177+ neutrophil effector program—defined by genes mediating degranulation, reactive oxygen species production, leukocyte transmigration, and neutrophil extracellular trap (NET) formation—with the transcription factor SPI1 implicated as a master regulator [35]. This discovery establishes a unifying framework connecting KD and MIS-C pathogenesis through shared neutrophil activation mechanisms.
4) T-cell remodeling and adaptive immune reprogramming
Beyond innate immunity, single-cell and integrative analyses increasingly highlight T-cell remodeling in KD. Enhanced activation of CD8+ cytotoxic T cells has been consistently observed [32], together with an imbalance between Th17 and regulatory T (Treg) cells. Most studies report an increase in IL-17A–producing Th17 cells accompanied by a relative reduction or functional impairment of Tregs [36], suggesting a shift toward a proinflammatory adaptive immune milieu. In a transcriptome-profiling study of KD coronary arteries, up-regulated pathways included T lymphocyte activation and type I interferon response, consistent with cytotoxic T-cell involvement [26]. In parallel, circulating levels of chemokines such as CXCL10 (IP-10) are elevated in acute KD, potentially promoting recruitment of effector T cells to inflamed vascular tissues [37]. Together, these findings suggest that, in addition to neutrophil-driven inflammation, adaptive immune reprogramming contributes to vascular pathology and potentially to repair processes depending on disease stage and treatment status.
5) Emerging spatial transcriptomics and vascular microenvironments
Spatial transcriptomic approaches in KD remain in their infancy, but proof-of-concept studies in murine models are emerging. A recent study employing spatial transcriptomics combined with scRNA-seq in a mouse vasculitis model revealed focal immune-cell infiltration and up-regulation of IL-1β/IL-18 signaling within vascular lesions [38]. Similarly, spatial transcriptomic analysis of coronary tissue from the Lactobacillus casei cell wall extract (LCWE) mouse model (GSE178799) demonstrated localized gradients of inflammatory transcripts, illustrating how spatially restricted immune–endothelial crosstalk may drive coronary artery injury.
Although human studies remain limited due to scarce coronary tissue availability, the ongoing development of high-resolution and minimally invasive spatial profiling technologies holds great promise for elucidating the vascular immune microenvironment in KD.
6) Integrative perspective and future directions
Collectively, bulk, single-cell, and spatial transcriptomic analyses converge on a model in which KD is driven by neutrophil hyperactivation, adaptive immune remodeling, and localized vascular inflammation. The conserved CD177+ neutrophil program establishes a mechanistic link between systemic cytokine responses and vascular injury, while insights into T-cell dynamics and nascent spatial mapping underscore the immunovascular microenvironment as a critical pathogenic nexus. Future work integrating transcriptomic data with immune receptor sequencing, proteomics, and epigenomics will help delineate cellular circuits underlying KD and advance precision immunomodulatory strategies.
3. Proteomics
1) Rationale — why proteomics matters in KD
Genomics and transcriptomics define susceptibility and transcriptional programs, but proteomics measures the actual effector molecules mediating inflammation, endothelial injury, and tissue remodeling. Mass-spectrometry (MS)-based discovery proteomics and affinity-based high throughput targeted proteomics platforms (e.g., Olink, SomaScan) now permit unbiased quantitation of hundreds-to-thousands of circulating proteins, enabling (1) identification of mechanistic effectors that bridge blood signatures to vascular pathology, (2) discovery of candidate biomarkers for diagnosis/IVIG response/CAL risk, and (3) evaluation of pathway activity for therapeutic targeting. Recent reviews summarize this shift toward MS- and affinity-based proteomics in pediatric inflammatory syndromes, including KD [39,40].
2) Acute-phase proteomic signatures: neutrophil and endothelial effectors
Unbiased proteomic analyses of KD plasma/serum repeatedly show up-regulation of neutrophil-derived proteins (S100A8/A9/A12, myeloperoxidase, neutrophil elastase) and acute-phase reactants (c-reactive protein, serum amyloid A), mirroring transcriptomic neutrophil signatures and supporting a central role for neutrophil activation in acute KD. Targeted/label-free MS and iTRAQ (isobaric tags for relative and absolute quantitation) studies have specifically identified S100A8/A9/A12 among top discriminating proteins which are linked to later coronary involvement in some cohorts [41].
Endothelial and coagulopathy-related proteins—including soluble adhesion molecules, von Willebrand factor, and serine protease inhibitor clade E member 1 (SERPINE1, also known as plasminogen activator inhibitor-1 [PAI-1])—are also elevated in acute KD, implicating early endothelial dysfunction and thrombogenic signaling. Notably, recent proteomic analyses identified SERPINE1 as a candidate predictor of CAL formation [42].
3) Proteomic correlates of IVIG response
Prediction of IVIG resistance remains a major clinical challenge. Comparative proteomic studies reveal that IVIG nonresponders exhibit higher pretreatment levels of neutrophil-derived inflammatory proteins (S100A8/A9, MRP8/14), proteases (neutrophil elastase), and complement/Fc-binding components—signatures consistent with exaggerated innate activation. Integrating proteomic markers with clinical variables significantly improves discrimination of IVIG nonresponse compared with clinical scores alone [41,43].
4) CAL-associated proteomic markers
Proteomic profiling implicates extracellular matrix-remodeling enzymes (MMP-8, MMP-9, cathepsins) and endothelial injury markers in CAL-positive patients, supporting a model of proteolytic degradation and aberrant angiogenic signaling in aneurysm formation. Persistent elevation of vascular injury-related proteins after IVIG treatment suggests incomplete resolution of endothelial inflammation and their potential utility as prognostic biomarkers or therapeutic targets [44,45].
5) Pathway and network interpretation
Pathway and network analyses of KD proteomes reveal three interacting functional modules: (1) Innate immune activation—neutrophil degranulation, complement activation, S100-driven inflammation. (2) Endothelial dysfunction and remodeling—adhesion molecules, coagulation regulators (SERPINE1), and MMP-mediated extracellular matrix degradation. (3) Metabolic rewiring—altered glycolytic and fatty acid oxidation enzymes linking immune-cell energetics to effector activity. These network hubs (e.g., S100 proteins, MMPs, SERPINE1) represent mechanistic candidates for targeted modulation and for development into multiplex biomarker panels [42,44].
6) Integration with other omics
Cross-omic comparisons reveal both concordant and discordant patterns between transcript and protein levels, underscoring the influence of posttranscriptional regulation and secretion dynamics. Proteogenomic approaches connect KD susceptibility loci (e.g., FCGR2A, ITPKC) to downstream protein networks, while combined proteome-metabolome analyses delineate coherent immune-metabolic phenotypes associated with IVIG resistance and vascular remodeling [29]. This integrative framework provides mechanistic links between genetic predisposition, cytokine signaling, and effector protein expression.
7) Emerging platforms and translational roadmap
Recent high-plex proteomic technologies are expanding translational potential in KD research.
The Olink platform identified IL-17A as a candidate biomarker differentiating KD from other febrile illnesses, highlighting an additional cytokine axis complementing neutrophil- and IL-1–dominated signatures [46]. Indeed, IL-17 is known to promote neutrophil-mediated immune responses [47]. Similarly, SomaScan proteomic profiling revealed differential abundance of hundreds of proteins across KD subgroups, capturing molecular and clinical heterogeneity within the disease [48]. Together, these technologies bridge proteomics and clinical translation, enabling high-throughput, standardized protein quantification. When integrated with genomic, transcriptomic, and metabolomic data, such proteomic frameworks can refine patient stratification, enhance biomarker-driven diagnostics, and accelerate the development of precision therapeutics for KD.
4. Metabolomics
1) Metabolomics as the functional readout
Metabolomics serves as the most proximate readout of cellular function, capturing the small-molecule substrates, intermediates, and products of biochemical pathways. In the context of KD, it provides a bridge from upstream genomic and transcriptomic perturbations to downstream effector metabolism, particularly in immune and vascular cells.
2) Lipid remodeling: LPC/LPE depletion and mechanistic hypotheses
One reproducible metabolomic observation in KD is decreased circulating lysophosphatidylcholine (LPC) and lysophosphatidylethanolamine (LPE) species in the acute phase [49,50]. Mechanistically, a plausible explanation is inflammation-driven activation of phospholipase A2 (PLA2), which hydrolyzes membrane phospholipids to generate lysophospholipids and free fatty acids (including arachidonic acid) that feed proinflammatory eicosanoid and LPA pathways [51,52]. Altered LPC/LPE pools can influence endothelial and immune-cell function directly—for example by modulating receptor signaling platforms, promoting leukocyte recruitment, and shifting the balance of pro- versus anti-inflammatory lipid mediators—as reviewed for LPC biology [53]. Taken together, LPC/LPE depletion in KD likely reflects both downstream consequences of oxidative/inflammatory lipid remodeling and upstream contributions to vascular inflammation, although direct demonstration of PLA2 overactivation in human KD remains to be established.
3) Amino acid and energy metabolism: acylcarnitines and tryptophan-kynurenine axis
Although studies on acylcarnitine changes in KD are limited, acylcarnitine dysregulation is a well-characterized marker of mitochondrial β-oxidation dysfunction in multiple disease contexts [54]. Plasma metabolomics in patients with systemic inflammation frequently reflect altered acylcarnitine species, suggesting immune bioenergetic stress in KD as well.
In parallel, perturbations in other amino acid metabolic pathways have been observed, notably in the arginine-nitric oxide (NO) pathway [55]. Disrupted arginine metabolism, which is pivotal for endothelial NO production, may contribute to endothelial dysfunction and vascular inflammation in KD, consistent with its vasculitic phenotype.
In addition, untargeted metabolomic profiling has demonstrated altered tryptophan metabolism in KD, with reduced tryptophan and increased kynurenine/kynurenic acid levels in patient cohorts [56]. This supports the involvement of the tryptophan-kynurenine axis, which is closely linked to cytokine-driven immune regulation through indoleamine 2,3-dioxygenase 1 (IDO1) activity. The exact role of this pathway in the pathogenesis of KD is yet to be determined since kynurenine is known to suppress inflammation and T-cell immune responses [57]. Increased levels of kynurenine in KD could be a counterregulatory response to increased inflammation.
Finally, bile acid metabolic alterations have been identified in KD, implicating dysregulation of the gut-liver-immune axis [58]. These findings raise the possibility that microbiome-derived metabolites modulate systemic inflammation, vascular injury, and treatment response variability.
4) Cross-study consistency and technical caveats
KD metabolomic studies employ various platforms (GCMS, LC-MS/MS, lipidomics pipelines, targeted acylcarnitine assays), and heterogeneity in sample type (serum vs. plasma), fasting status, timing relative to fever onset or IVIG, and normalization strategies introduces variability. Nonetheless, consistent pathway-level themes—lipid remodeling, mitochondrial stress, tryptophan metabolism—emerge across studies, reinforcing their biological relevance. Future work must standardize protocols, ensure replication in independent cohorts, and carefully control confounding factors.
5) Clinical implications, limitations, and future directions
Metabolomic signatures hold promise as early biomarkers for IVIG resistance and CAL risk, and may suggest adjunctive metabolic therapies (e.g., antioxidant, mitochondrial modulators, IDO pathway modulators). However, present evidence is still exploratory, with most studies constrained by small sample sizes, single-center design, and lack of prospective validation [59].
Looking ahead, a major frontier is single-cell metabolomics: just as scRNA-seq resolved cellular heterogeneity, future single-cell or spatial metabolomic methods could directly quantify metabolic states in hyperactivated neutrophils, IL-1 high monocytes, or endothelial cells. Such technology would enable linking cellular bioenergetics to transcriptomic and proteomic phenotypes in KD. Integrative metabolomics combined with immune receptor sequencing, proteomics, and transcriptomics can delineate causal pathways from genotype to phenotype, refine mechanistic targets, and guide precision immunometabolic therapy in KD.
5. Epigenomics
Epigenetic mechanisms—including DNA methylation, noncoding RNAs (microRNAs [miRNAs], long noncoding RNAs [lncRNAs], and exosomal RNAs), and chromatin-level regulation—provide a dynamic interface between genetic susceptibility, environmental exposures (including infectious triggers), and immune activation in KD. Unlike static genetic variants, epigenetic modifications are reversible and responsive to inflammatory cues, thereby shaping both acute immune responses and longer-term vascular outcomes.
1) DNA methylation: dominant and reproducible epigenetic alterations
Genome-wide methylation studies consistently demonstrate widespread DNA methylation changes during acute KD, particularly within immune-related loci such as FCGR2A/B, FCER1A, and inflammasome-associated genes (e.g., NLRC4, NLRP3) [60,61]. Hypomethylation of neutrophil and inflammasome genes correlates with heightened innate immune activation, whereas hypermethylation at T-cell regulatory loci parallels transcriptomic evidence of lymphocyte suppression [62,63].
Altered expression of DNA methyltransferases (DNMT1, DNMT3A) has been linked to CAL formation [64], suggesting transient global hypomethylation driven by systemic inflammation. Cytokine signaling pathways—particularly IL-6–STAT3—may directly suppress DNMT activity, reinforcing a cytokine-epigenetic feedback loop in acute KD [65].
2) Noncoding RNAs: circulating and cell-associated regulatory signals
Changes in miRNA expression represent another reproducible epigenetic feature of KD. Multiple studies report differential expression of miR-223, miR-145, miR-155, and miR-92a during the acute phase, with partial normalization after IVIG treatment. These miRNAs are implicated in toll-like receptor/nuclear factor kappa B signaling, neutrophil and monocyte activation, endothelial responses, and treatment responsiveness [61,66].
Exosomal miRNAs and lncRNAs have emerged as potential mediators of immune-vascular communication, acting in a paracrine manner to regulate endothelial activation, leukocyte adhesion, and vascular permeability [67,68]. However, heterogeneity in profiling platforms and limited cohort sizes currently restrict their immediate clinical utility, emphasizing the need for standardized and multicenter validation.
3) Histone modifications and chromatin remodeling (emerging layer)
Evidence for histone-level regulation in KD remains limited and largely exploratory. Preliminary reports suggest altered histone acetylation (H3K27ac) and methylation (H3K4me3) at inflammatory loci, implying that chromatin accessibility may contribute to sustained transcriptional activation beyond the acute cytokine surge [69,70]. Advanced approaches such as ChIP-seq, CUT&Tag, and scATAC-seq hold promise for resolving cell-type–specific chromatin landscapes in immune and vascular compartments, but currently represent an emerging research direction rather than an established pathogenic framework.
4) IVIG as an epigenetic modulator
IVIG therapy induces substantial epigenetic remodeling, in some cases exceeding changes observed at disease onset [60,63]. These include normalization of hypomethylated inflammatory loci and partial reversal of neutrophil-associated epigenetic signatures, suggesting that IVIG exerts durable immunomodulatory effects partly through epigenetic reprogramming rather than solely through passive immune complex neutralization.
5) Translational perspective and limitations
Integrative analyses indicate that epigenetic alterations may stabilize acute immune dysregulation into longer-lasting inflammatory or vascular phenotypes. Differentially methylated regions and circulating miRNA panels therefore represent candidate biomarkers for disease activity, IVIG response, and CAL risk. Nevertheless, current evidence is constrained by bulk-tissue analyses, small sample sizes, and limited longitudinal sampling. Future priorities include single-cell or spatial epigenomic profiling, pre- and post-IVIG longitudinal designs, and functional validation using epigenome-editing or induced pluripotent stem cell-derived vascular and immune models. At present, epigenomics in KD primarily provides mechanistic and hypothesis-generating insights that complement genomic and transcriptomic findings.
6. Immunomics
Immunomics—encompassing immune-cell composition, receptor repertoires (TCR/BCR), and soluble immune mediators at high dimensionality—provides an integrated framework to decipher the immune dysregulation underlying KD. High-dimensional technologies such as single-cell transcriptomics with paired TCR/BCR sequencing, mass cytometry (CyTOF), and cytokine proteomics have revealed both conserved and patient-specific immune states that connect systemic inflammation to vascular injury, IVIG resistance, and treatment response heterogeneity.
1) Innate immune drivers: neutrophils and monocytes
Across single-cell and bulk datasets, neutrophil hyperactivation is a consistent hallmark of acute KD. Expansion of CD177+ neutrophil subsets with degranulation, reactive oxygen species/neutrophil extracellular traps (ROS/NET) formation, and endothelial-homing signatures defines a shared neutrophil effector program observed in KD and MIS-C. This program attenuates after IVIG treatment and correlates with clinical inflammation and coronary artery involvement [35].
Monocytes, particularly classical CD14++CD16− and CD14+CD16+ intermediate subsets, display proinflammatory and inflammasome-related transcriptomes with diminished antigen-presentation programs, indicating a shift toward innate effector function. In infants with KD, scRNA-seq and flow-cytometric studies have shown expansion of SELL+CD14+CD16− classical monocytes with neutrophil-activating gene signatures, as well as an increase in CD14+CD16+ intermediate monocytes during the acute phase of disease [71-73]. Intercellular network analyses from single-cell PBMC data position these monocytes as central communication hubs linking neutrophils, endothelial cells, and adaptive lymphocytes [32,74].
Collectively, these innate immune programs provide both dynamic biomarkers for monitoring disease activity and IVIG response, and a clear mechanistic rationale for targeting innate cytokines (e.g., IL-1) and neutrophil effectors in refractory KD [35,75].
2) Adaptive immunity: T-cell remodeling and TCR features
Single-cell analyses have uncovered extensive T-cell remodeling in KD, with perturbed CD4/CD8 ratios, expansion of cytotoxic CD8+ T cells, and disrupted Th17/Treg balance. Paired scRNA–TCR sequencing demonstrates clonal expansions and a higher-than-expected frequency of dual-TCR T cells, which express two different TCRs, suggesting antigen-driven or superantigen-like responses in subsets of patients [32,76]. These findings implicate antigen-specific T-cell activation as a key driver of vascular inflammation, while interindividual heterogeneity underscores the importance of immune context and genetic background.
3) B cells, plasmablasts and antibody responses
Proteomic and repertoire analyses show transient plasmablast expansion during acute KD and a polyclonal activation pattern across B-cell receptors. Although convergent clonotypes have not been consistently observed, some studies have reported biased usage of immunoglobulin heavy chain variable gene families.
The potential contribution of anticytokine or antiendothelial autoantibodies—either endogenously produced or passively transferred via IVIG—remains under investigation. Understanding these transient humoral phenomena may help explain variable therapeutic responses and disease phenotypes [77].
4) Soluble immunomics: cytokine and proteomic signatures
High-plex proteomic profiling consistently highlights IL-1 and IL-6 pathway activation in acute KD, along with emerging evidence for IL-17 family cytokines as potential markers of IVIG resistance or coronary risk. Although Th17 cells have been implicated, recent single-cell studies point to innate lymphoid cells as rapid IL-17 producers that bridge innate and adaptive inflammation [78,79].
Elevated IL-1/IL-6 signaling provides a clear rationale for cytokine-targeted therapies, such as anakinra or tocilizumab, though population heterogeneity and sampling variability currently limit universal biomarker application [80,81].
5) Immune receptor repertoires and antigen specificity
Comprehensive TCR/BCR repertoire sequencing in KD remains in early stages but is rapidly advancing with integrated scRNA+TCR/BCR pipelines. Early findings indicate clonal T-cell expansions and biased V-gene usage, supporting an antigen-driven response hypothesis. However, no single public antigenic signature has yet been identified. Rigorous methodology and large, ethnically diverse cohorts will be essential for reproducible discovery [76,82].
6) Multiomic integration and translational implications
Integrative analyses linking immune-cell transcriptomes, cytokine proteomics, and metabolic signatures have started to map the molecular cascade from cytokine activation (e.g., IL-1 → kynurenine pathway induction → endothelial dysfunction) to vascular injury [33].
Composite immune risk scores, incorporating cell-subset frequencies, cytokine profiles, and receptor clonality, show promise for predicting IVIG resistance and CAL risk [33,35]. To ensure clinical utility, these findings must be validated prospectively across diverse populations, with standardized sample timing, paired multi-omic profiling, and harmonized clinical metadata (disease severity, treatment regimens, coronary imaging outcomes). Immunomics thus bridges cellular mechanisms with clinical translation, offering both diagnostic precision and targeted therapeutic guidance for KD [75,83].
7. Microbiomics
Microbiomics examines whether and how microbial communities at mucosal surfaces—primarily the gut and upper airway—are associated with KD susceptibility, immune activation, and vascular injury. At present, evidence from human studies largely supports association rather than direct causation; however, converging observational and experimental findings suggest that microbial dysbiosis and microbe-derived metabolites may modulate immune and vascular responses relevant to KD pathophysiology.
1) Clinical microbiome studies – associative evidence
Case-control and longitudinal studies using 16S rRNA sequencing or shotgun metagenomics have reported alterations in the gut microbiota during the acute phase of KD compared with convalescence or healthy states. These changes are variably associated with markers of systemic inflammation and with alterations in microbial metabolites, including short-chain fatty acids and bile acids, linking microbial metabolism to host immune activation.
Studies of the nasopharyngeal and oral microbiome have similarly described compositional differences in KD, suggesting a potential contribution of upper-airway microbial communities to immune triggering. However, findings across human cohorts are heterogeneous and largely correlative, with substantial variability related to population, sampling site, timing, and analytical methods.
Overall, current clinical microbiome data support an association between dysbiosis and immune activation, rather than a definitive causal role in KD pathogenesis.
Details of reported taxa are summarized in Supplementary Table 1.
2) Insights from animal models
Mechanistic support for microbiome involvement in KDlike vasculitis primarily derives from the LCWE mouse model. In this system, coronary arteritis is driven by IL-1R/MyD88-dependent innate immune pathways, and experimental manipulation of gut microbiota—through antibiotics or probiotic reconstitution—modulates disease severity [23] These studies provide proof-of-principle that microbial composition can influence immune tone and vascular inflammation, although direct extrapolation to human KD remains limited.
3) Putative immune-metabolic mechanisms
Proposed mechanisms linking microbes to KD include immune activation by microbial products and modulation of host immunity by microbial metabolites. Microbiome-derived bile acids, short-chain fatty acids, and tryptophan metabolites can influence IL-1 and IL-6 signaling, immune-cell metabolism, and epigenetic regulation. In addition, increased intestinal permeability during systemic inflammation may enhance exposure to microbial components, promoting neutrophil recruitment and vascular inflammation. Together, these observations support a gut-immune-vascular axis, best interpreted as a modulatory rather than initiating pathway in KD.
4) Limitations and future directions
Microbiome studies in KD are constrained by small sample sizes, regional and methodological heterogeneity, and limited longitudinal data. Importantly, a recent Mendelian randomization analysis did not demonstrate a clear causal relationship between gut microbiota and KD, underscoring the complexity of host-microbe interactions.
Future progress will require well-powered, longitudinal, multiomic studies integrating microbiome, metabolomic, and host immune profiling, alongside functional validation in experimental models. In this context, microbiomics is best viewed as a contextual and modulatory layer that may refine immune stratification or risk prediction, rather than as a primary causal driver of KD.
Integrated multiomics: the molecular cascade of KD
Integrative multiomics seeks to reconstruct the molecular cascade linking inherited susceptibility and environmental triggers to immune activation and endothelial injury, thereby improving biological understanding of IVIG resistance and coronary outcomes. This integrative view enables (1) prioritization of plausible causal genes at GWAS loci, (2) mapping of regulatory effects to specific cell types, (3) elucidation of cross-layer interactions (e.g., cytokine → metabolism → epigenetic remodeling), and (4) development of composite signatures that may support future risk stratification and monitoring [84,85].
A variety of integrative analytical frameworks have been applied in KD to link multiomics data with clinical phenotypes (Supplementary Table 2). In brief, these approaches aim to (1) identify shared inflammatory or immune-metabolic axes across omics layers, (2) derive composite molecular signatures associated with IVIG response or coronary outcomes, and (3) map molecular signals to specific immune or vascular cell populations [84,86-89].
1. Convergent molecular axes in KD
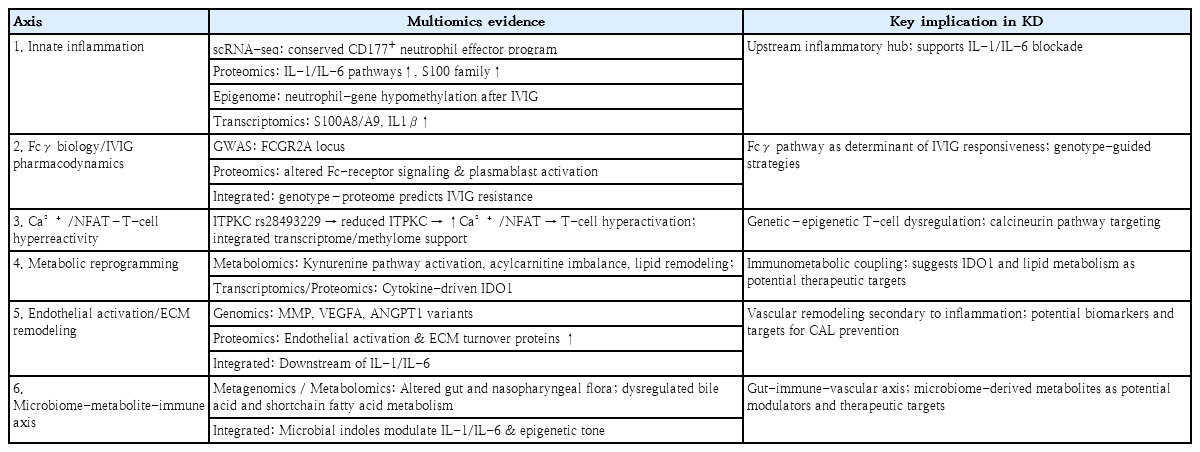
Integrative studies consistently converge on six key mechanistic axes (Table 1): (1) Innate inflammation dominated by IL-1/IL-6-driven neutrophil programs [29,87], (2) Fcγ receptor signaling modulating IVIG pharmacodynamics [8,90], (3) Ca²+/NFAT pathway variants (e.g., ITPKC) linking genetic susceptibility to T-cell activation [2,91] (4) Metabolic rewiring involving kynurenine and lipid remodeling under cytokine-driven mitochondrial stress [56,92], (5) Endothelial and ECM remodeling predisposing to coronary aneurysm formation [93,94], and (6) Microbiome-metabolite-immune crosstalk connecting environmental triggers to systemic inflammation and vascular outcomes [58]. These axes collectively depict a systems-level cascade from immune activation to vascular pathology.
Representative multiomics integration strategies and applications
2. Integrative framework for mechanistic and therapeutic translation
Recent multiomics research reframes KD as a coordinated immune-metabolic-vascular disorder rather than a purely idiopathic vasculitis. Functional variants such as ITPKC and FCGR2A remain key genetic anchors, shaping T-cell calcium signaling and Fcγ biology respectively [2,8].
Emerging single-cell and factor-analysis approaches are now mapping these cross-layer effects onto defined immune and endothelial populations, uncovering IL-1/IL-6-driven transcriptional programs relevant to neutrophil and vascular activation [86,95].
Epigenetic remodeling appears to underlie disease persistence: DNA methylation and chromatin accessibility changes after acute KD and IVIG have been linked to CAL formation and DNMT downregulation, suggesting that transient cytokine cues may imprint long-lasting inflammatory or vascular remodeling signatures [61,64].
Cross-omic analyses also highlight coupling between cytokine signaling and metabolic reprogramming, including activation of the tryptophan-kynurenine pathway and acylcarnitine perturbations indicative of mitochondrial stress—mechanisms plausibly connecting immune activation to endothelial injury and IVIG resistance [54,56].
Microbiome studies add an environmental dimension: TCR repertoire and scRNA/TCR-seq analyses support superantigen-like microbial activation in subsets of KD, while integrated microbiome-metabolome analyses have linked altered bile acid and tryptophan-derived metabolite pools to inflammatory phenotypes [76,96]. Although current findings remain preliminary, they collectively support a gut-microbe-metabolite-immune axis influencing disease severity and coronary outcomes.
Methodologically, early multiomic classifiers combining transcriptomic, proteomic, and metabolomic features have shown potential for improving prediction of IVIG resistance and coronary complications in research settings. However, larger, multiancestry, longitudinal studies integrating host genetics, immune states, and microbial ecology will be essential to establish causality and enable precision-guided therapies [86,95].
Taken together, converging evidence supports a unified molecular cascade in KD: innate cytokine activation (IL-1/IL-6) drives neutrophil effector expansion and metabolic stress, leading to epigenomic remodeling, endothelial activation, and ECM dysregulation that culminate in coronary pathology. Better understanding of this cascade would provide therapeutic interventions, such as IL-1/IL-6 blockade, metabolic modulation, and Fcγ-axis targeting, laying the groundwork for future precision immunotherapy in KD [35,56].
Clinical translation: what is actionable now, what remains investigational
1. Near-term opportunities (requires prospective validation)
First, multimarker blood-based panels integrating inflammatory proteins and transcripts may improve risk stratification for IVIG resistance beyond existing clinical scores, particularly outside Japanese populations. Second, longitudinal profiling across standardized time points (pre-IVIG, 36- to 48-hour post-IVIG, and early convalescence) can support disease monitoring and early identification of persistent vascular inflammation. Third, combining molecular features with echocardiographic data may enable better prediction of coronary outcomes, including early CAL progression.
2. Research-stage insights (not yet clinically deployable)
Mechanistic findings from spatial omics, microbiome studies, rare-variant discovery, and epigenetic/histone-level regulation remain largely hypothesis-generating due to limited replication, scarce tissue availability, and methodological heterogeneity. These layers are valuable for identifying causal circuits and therapeutic targets but should not be interpreted as ready for routine clinical decision-making.
3. Key barriers to implementation
Major barriers include small and heterogeneous cohorts, variability in sample type and timing, confounding by treatment, lack of external validation, and challenges in converting high-dimensional signatures into standardized, cost-effective assays with rapid turnaround. Harmonized, longitudinal, multiancestry cohorts with shared protocols and clinically meaningful endpoints (IVIG response, CAL incidence/progression, long-term cardiovascular outcomes) are essential to bridge discovery and practice.
Conclusion
Multiomics studies have shifted the understanding of KD from a single-layer vasculitis to a network disease shaped by coordinated disturbances across genetic, immunologic, metabolic, and vascular compartments. Importantly, independent omics layers consistently highlight convergent mechanistic axes—including an IL-1/IL-6–driven neutrophil inflammatory hub (transcriptomics/proteomics), Fcγ-receptor signaling influencing IVIG pharmacodynamics (genomics/proteomics), Ca²+/NFAT-dependent immune activation (genomics/transcriptomics), and endothelial/ECM remodeling associated with coronary pathology (transcriptomics/proteomics). This cross-layer convergence strengthens biological plausibility and provides a coherent framework for interpreting clinical heterogeneity such as IVIG resistance and CAL risk.
At the same time, most multiomics findings in KD remain at the discovery-to-early validation stage, and translation into routine clinical tools is still limited. Moving forward, the most realistic near-term clinical opportunities include improved risk stratification (e.g., identifying patients at higher risk of IVIG resistance or coronary complications), disease monitoring through biologically informed signatures, and long-term outcome prediction using standardized longitudinal sampling. Achieving these goals will require harmonized, multiancestry cohorts; standardized sampling across disease phases; transparent analytical pipelines; and prospective validation demonstrating added value over conventional clinical and laboratory markers. With these steps, multiomics can provide a practical translational roadmap—linking mechanism-based insights to clinically meaningful endpoints—while avoiding overstatement of immediate diagnostic or therapeutic applicability.
Supplementary materials
Supplementary Tables 1-2 are available at https://doi.org/10.3345/cep.2025.02901.
Lifestyle modification program: stages, goals, and specialist roles
Lifestyle modification program: stages, goals, and specialist roles
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author contribution
Conceptualization: JGA, IK; Data curation: JGA; Formal analysis: JGA; Methodology: JGA, IK; Project administration: JGA; Visualization: JGA; Writing - original draft: JGA; Writing - review & editing: JGA, IK