High-Risk CCL14 lineage drives severe Mycoplasma pneumoniae pneumonia in children: a study in central China
Article information

Abstract
Background
The post–coronavirus disease 2019 (COVID-19) pandemic resurgence of Mycoplasma pneumoniae (MP) infections, particularly in China, underscores the need to understand the drivers of disease severity.
Purpose
This study aimed to evaluate the association between predominantly circulating MP strains and disease severity in Henan Province, central China.
Methods
We integrated the clinical data of 3,060 pediatric patients and analyzed the epidemiological characteristics of MP pneumonia (MPP) in the Henan region in 2020–2024. Bronchoalveolar lavage fluid collected from 137 patients was analyzed using multilocus sequence typing and multilocus variable number tandem-repeat analysis to investigate the correlation between genotype and clinical outcome.
Results
A significant post-COVID MPP outbreak was predicted in 2023. Genotyping revealed the cocirculation of 2 major genotypes: the prevalent ST3 (57.7%, severe ratio [31.6%]) and the less common but highly virulent ST14 (26.3%, severe ratio [72.2%]). Phylogenetic clustering confirmed ST14/3-5-6-2 as part of a broader hypervirulent lineage, common cluster label 14 (CCL14), which was significantly associated with disease severity (χ2=19.39; P<0.001). Patients infected with CCL14 strains exhibited a distinct hyperinflammatory profile marked by elevated D-dimer levels, complement C4 levels, and platelet count. The mutation ratio of macrolide resistance sites in the MP strains from Henan Province was approximately 75%.
Conclusion
Our findings identified the CCL14 lineage as a key driver of disease severity in macrolide-resistant pediatric MPP in Henan Province, China. This under scores the importance of integrating molecular surveillance with clinical monitoring to mitigate disease burden.
Key message
Question: Are the predominant circulating strains of Mycoplasma pneumoniae associated with disease severity and clinical indicators?
Finding: The common cluster label 14 (CCL14) lineage was a high-risk clone circulating in central China that demonstrated a strong association between severe pediatric pneumonia and a distinct hyperinflammatory profile.
Meaning: Targeted molecular surveillance of the CCL14 lineage may facilitate early risk stratification and guide clinical management to reduce the burden of severe disease.
Graphical abstract. CCL, common cluster label; MP, Mycoplasma pneumoniae; MLVA, multilocus variablenumber tandem-repeat analysis; MLST, multilocus sequence typing; D-Di, D-dimer.
Introduction
Mycoplasma pneumoniae (MP) is a leading cause of community-acquired pneumonia (CAP) in children, accounting for approximately 40% of cases [1-3]. Although Mycoplasma pneumoniae pneumonia (MPP) is generally self-limiting, it may in certain cases progress to severe or refractory forms of the disease, resulting in an unfavorable prognosis [4]. It is also accompanied by extrapulmonary complications such as myocarditis and hepatitis [5] and may even lead to fatal outcomes [6].
MP infection occurs year-round across diverse global climates and is characterized by cyclical epidemics every few years [7]. During epidemic phases, MP infections often begin in early autumn and are more frequent in dry and cold climates, facilitating large-scale outbreaks throughout autumn and winter [8]. Post-COVID-19 pandemic, China has experienced a notable resurgence in pediatric MP infections. By late 2023, MP detection rates in pediatric CAP patients surged to 60%–70%, a figure substantially higher than pre-pandemic (~30%) and during-pandemic (10%–20%) levels [9]. Furthermore, this trend has resonated globally [10,11].
Molecular typing is crucial for MP epidemiological investigation. While P1 typing was the first method used and multilocus variable-number tandem-repeat analysis (MLVA) is widely applied for initial screening, multilocus sequence typing (MLST) provides superior discriminatory power [12,13]. Nevertheless, MLST is more costly and time-consuming. Given that MLVA offers complementary value for rapid surveillance and outbreak tracing, we employed both methods complementarily to obtain a comprehensive molecular epidemiological profile.
At present, genotyping data on MP strains circulating in Henan Province remains scarce. As a populous region with dry, cold winters that facilitate MP transmission, conducting epidemiological surveillance and genotyping research here is highly representative and holds significance for public health. Furthermore, different genotypes may influence bacterial virulence, transmissibility, and antibiotic resistance, potentially accounting for the observed variations in clinical manifestations and patient prognoses [14-16]. Despite this potential link, the genotypic landscape of MP in Henan and its correlation with clinical outcomes remain largely unexplored.
To address this gap, this study investigated the epidemiological characteristics of MP infections from 2020 to 2024. MLST and MLVA typing methods were employed in a complementary manner to conduct genotyping analysis on MP strains circulating in central China. By integrating patient clinical data, the associations between predominant circulating strains and disease severity, as well as clinical indicators, were systematically examined.
Methods
1. Study participants and design
A total of 3,060 pediatric patients diagnosed with MPP and hospitalized at Henan Children’s Hospital between June 2020 and May 2024 were included in this study. Comprehensive clinical data were extracted from the electronic medical record system, including demographic characteristics (age and sex), vital signs, comorbidities, laboratory test results, and imaging findings.
For the molecular genotyping component of this study, we specifically analyzed bronchoalveolar lavage fluid (BALF) samples. BALF was chosen as it provides a direct specimen from the site of infection, thereby offering a more accurate representation of the causative strain compared to upper respiratory tract samples, which may contain colonizing organisms. During this period, bronchoscopy with BALF sampling was clinically indicated for 275 patients based on the criteria. Finally, considering the sample quality, the collection time of the samples, and the coverage of the disease severity spectrum, and after excluding cases of coinfections, 137 MPP patients were selected for inclusion in the genotyping analysis. All participants underwent BALF testing based on clinical indications during the study period. This study was conducted in accordance with the Declaration of Helsinki, approved by the Ethics Committee of Zhengzhou University (No. ZZUIRB2024-204), and written informed consent was obtained from both the participants and their parents/legal guardian (s).
2. Diagnostic criteria and the definition of severe cases
The diagnostic criteria for MPP, the definition of severe cases, and the indications for performing bronchoscopy were implemented with reference to the "Guidelines for the Diagnosis and Treatment of M. Pneumoniae Pneumonia in Children (2023 Edition)." [17] The diagnostic criteria were: (1) a confirmed diagnosis of MPP in patients under 18 years of age; (2) presence of clinical manifestations such as cough, fever, tachypnea, and abnormal breath sounds; (3) typical imaging findings consistent with MPP, including interstitial infiltrates, segmental and lobar consolidations, as well as enlargement of the hilar lymph nodes; (4) availability of complete case records that fulfill the informational requirements of the study; (5) exclusion of patients with tracheal or pulmonary dysplasia and other congenital conditions. If one or more additional pathogenic infections are identified simultaneously through nucleic acid and antibody tests, the case is classified as a coinfection.
Pediatric patients were classified as having severe MPP if they met one or more of the following criteria during hospitalization: (1) persistent high fever (≥39°C for ≥5 days, or any fever lasting ≥7 days without improvement); (2) significant respiratory distress (dyspnea, hypoxemia with SpO2 ≤93% on room air); (3) radiological evidence of extensive lobar consolidation (involving ≥2/3 of a lobe or multiple lobes) or rapid progression (>50% increase in infiltrates within 48 hours); (4) development of pulmonary or extrapulmonary complications; (5) serum inflammatory markers, such as C-reactive protein, lactate dehydrogenase, and D-dimer, show a significant elevation.
3. Standardized clinical management pathway
All patients received standard macrolide treatment after being diagnosed with MPP. Systemic corticosteroids were added for cases meeting the severity criteria. Bronchoscopy with BALF sampling was not performed initially but was indicated only after ≥72 hours of this standard therapy in cases with suspected clinical or radiological progression. This ensured that genotypes were obtained under uniform therapeutic conditions, minimizing confounding by differential treatment exposures prior to sampling.
4. DNA extraction
To genotype MP isolates from patient samples, total DNA was extracted from BALF using a modified phenolchloroform method. The detailed extraction protocol followed established procedures as described in previous studies [18]. The concentration and purity of the extracted DNA met the quality standards necessary for downstream molecular analyses.
5. MP genotyping, macrolide resistance mutation analysis and DNA sequencing
MLVA typing was performed by amplifying and analyzing 4 variable-number tandem-repeat loci—Mpn13, Mpn14, Mpn15, and Mpn16. The MLVA type was defined by the specific combination of repeat numbers at these 4 loci, expressed in the format Mpn13–Mpn14–Mpn15–Mpn16 [19]. For MLST typing, sequence types were determined by sequencing 8 housekeeping genes: ppa (MPN528), pgm (MPN628), gyrB (MPN003), gmk (MPN246), glyA (MPN576), atpA (MPN600), arc (MPN307), and adk (MPN185) [20]. The specific allelic profile can be retrieved from the PubMLST website. All primers used in both genotyping methods were designed and applied in accordance with previously established protocols [19,20].
Primers (23rRNA-F 5’-GTCTCGGCTATAGACTCGTG-3’ and 23rRNA-R 5’-GCTACAACTAGCATAAG-3’) were employed to amplify the fragment of 23S rRNA nucleotides spanning positions 1997–2707. Subsequently, the amplified molecules were subjected to sequencing to determine whether mutations had occurred at the macrolide-resistant sites located at positions 2063, 2064, and 2617.
In this study, polymerase chain reaction (PCR) was used to amplify specific gene sequences for molecular genotyping of MP. Amplification was carried out in a T100 Thermal Cycler (Bio-Rad, USA) under the following conditions: initial denaturation at 95°C for 3 minutes; 35 cycles of denaturation at 94°C for 15 seconds, annealing at 50°C for 15 seconds, and extension at 72°C for 1 minute; followed by a final extension at 72°C for 5 minutes. The PCR products were separated by electrophoresis on a 1% agarose gel containing 0.5 μg/mL ethidium bromide, using a DYCP-31DN horizontal electrophoresis system (Liuyi, China). Amplification efficiency of the target gene fragments was assessed by visualizing the bands under ultraviolet light. Sequencing was performed by Sangon Biotech Co., Ltd. using the Sanger method. We received and reviewed the sequencing chromatograms for each sample to ensure: (1) clear, single-peak signals (high signal-to-noise ratio) across the target region, and (2) unambiguous base calling at the critical positions. Only sequences passing this manual quality check were used for mutation analysis.
6. Molecular clustering and common cluster label assignment
Population structure was analyzed using the goeBURST algorithm (fully iterated, single-locus variant [SLV] priority) on the PHYLOViZ Online 2.0 platform. The resultant minimum spanning tree (MST) was used to define clonal complexes and assign common cluster labels (CCLs). Sequence types were considered linked and grouped into the same CCL if they were SLVs or double-locus variants, i.e., differing at one or 2 of the 8 MLST loci, respectively. The largest interconnected group within the MST was designated as the primary CCL.
7. Phylogenetic analysis
To contextualize our local strains within a global framework, we retrieved MLST sequences of prevalent MP strains from Japan, the United Kingdom, and the United States from the PubMLST database (https://pubmlst.org/mpneumoniae). The sequences were categorized according to their regional prevalence, and representative sequences from each area were selected. These sequences, along with the MLST sequences obtained from clinical patient samples in this study, were used to construct a phylogenetic tree for evolutionary analysis using Molecular Evolutionary Genetics Analysis (ver. 11, MEGA Software, USA).
8. Statistical analysis
This study utilized IBM SPSS Statistics ver. 25.0 (IBM Co., USA) for statistical analysis. The normality of continuous variables was assessed using the Shapiro-Wilk test. Based on the normality test results, quantitative data conforming to a normal distribution are presented as mean±standard deviation and compared using the independent samples t test, while data not conforming to a normal distribution are presented as median with interquartile range and compared using the Mann-Whitney U test. Categorical variables were analyzed with the chi-square test and Fisher exact test. To identify independent risk factors for severe MPP, multivariable logistic regression analysis was performed. Variables were selected based on clinical relevance and findings from univariate analyses, all variables were forced into the model for adjustment. Results are presented as adjusted odds ratios (aOR) with 95% confidence intervals (CIs). Model fit was assessed using the Hosmer-Lemeshow test. All figures were generated using GraphPad (Prism ver. 9, GraphPad Software Inc., USA). A P value <0.05 was considered statistically significant.
Results
1. Epidemiological characteristics of MPP in hospitalized children
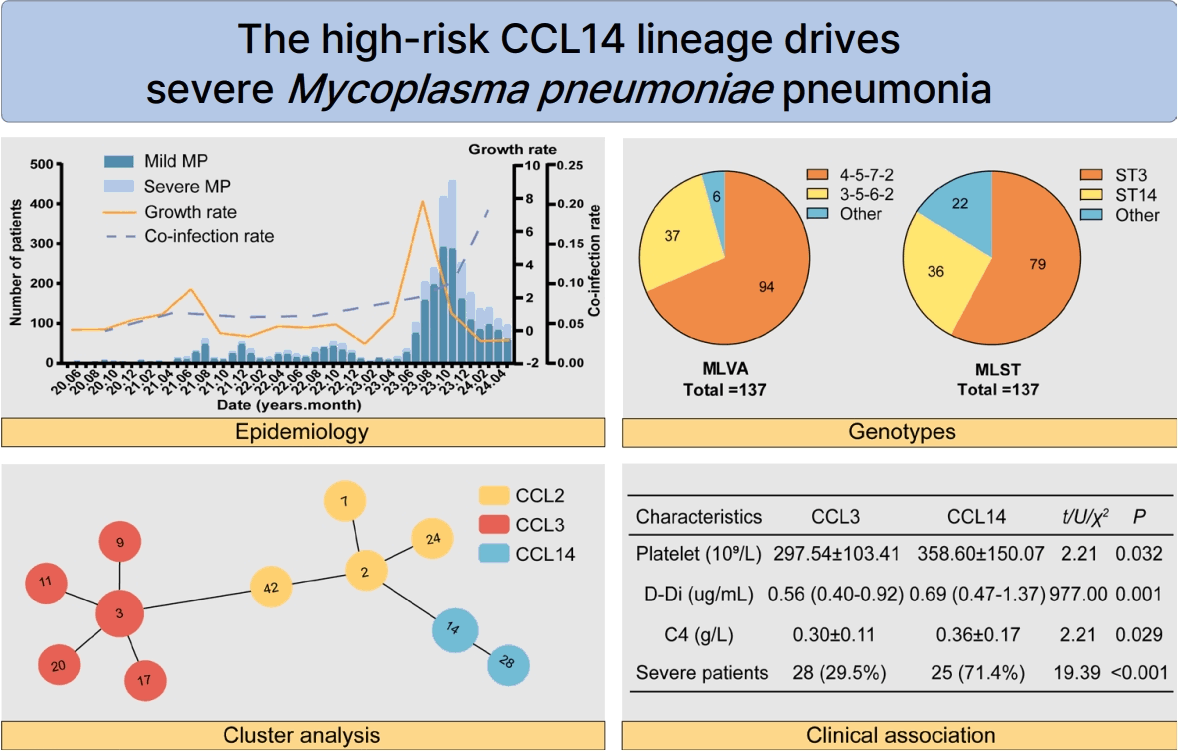
A total of 3,060 pediatric patients diagnosed with MPP who were treated at Henan Children's Hospital between June 2020 and May 2024 were included in this study. Prior to April 2021, the number of hospital admissions remained low, with both the total case count and growth trends exhibiting relative stability. Beginning in April 2021, a moderate increase in cases was observed, peaking in July of that year. Over the subsequent 2 years, the growth ratio gradually declined and fluctuated within a narrow range. A significant outbreak occurred in August 2023, characterized by a sharp rise in MP infections and a notable increase in the coinfection ratio. Throughout the study period, the proportion of severe cases varied from 0% to 44%, and the overall severity ratio of MPP in the population stood at 32.1% (982 of 3,060) (Fig. 1A).
Epidemiological characteristics of MPP in hospitalized children during and after coronavirus disease 2019 pandemic restrictions in Henan, China, from June 2020 to May 2024. (A) Temporal trends of hospitalized MPP cases among children in Henan Province, China, from June 2020 to May 2024. (B) Distribution of male and female pediatric patients diagnosed with MPP across different age groups and the proportion of severe cases. (C) Number of pediatric patients diagnosed with MPP infected by various pathogens across different age groups along with the associated coinfection ratios. MPP, Mycoplasma pneumoniae pneumonia; MP, Mycoplasma pneumoniae; ADV, adenovirus; SPN, Streptococcus pneumoniae; HI, Haemophilus influenzae; INFA, influenza A virus; RSV, respiratory syncytial virus.
In terms of age and sex distribution, the highest number of pediatric cases was observed in the 6 to 7-year-old cohort, which included a total of 826 individuals. Male patients outnumbered females across all age groups. The severity ratio increased with age, reaching its peak in the 10- to 11-year-old group (Fig. 1B). The number of coinfected children also rose with age, with the highest incidence observed in the 2- to 3-year-old group. However, beyond this age group, the ratio of coinfection demonstrated a declining trend. Among all age groups, adenovirus emerged as the most prevalent pathogen associated with coinfections (Fig. 1C).
2. Coinfection patterns among children with MPP in Henan Province, China from June 2020 to May 2024
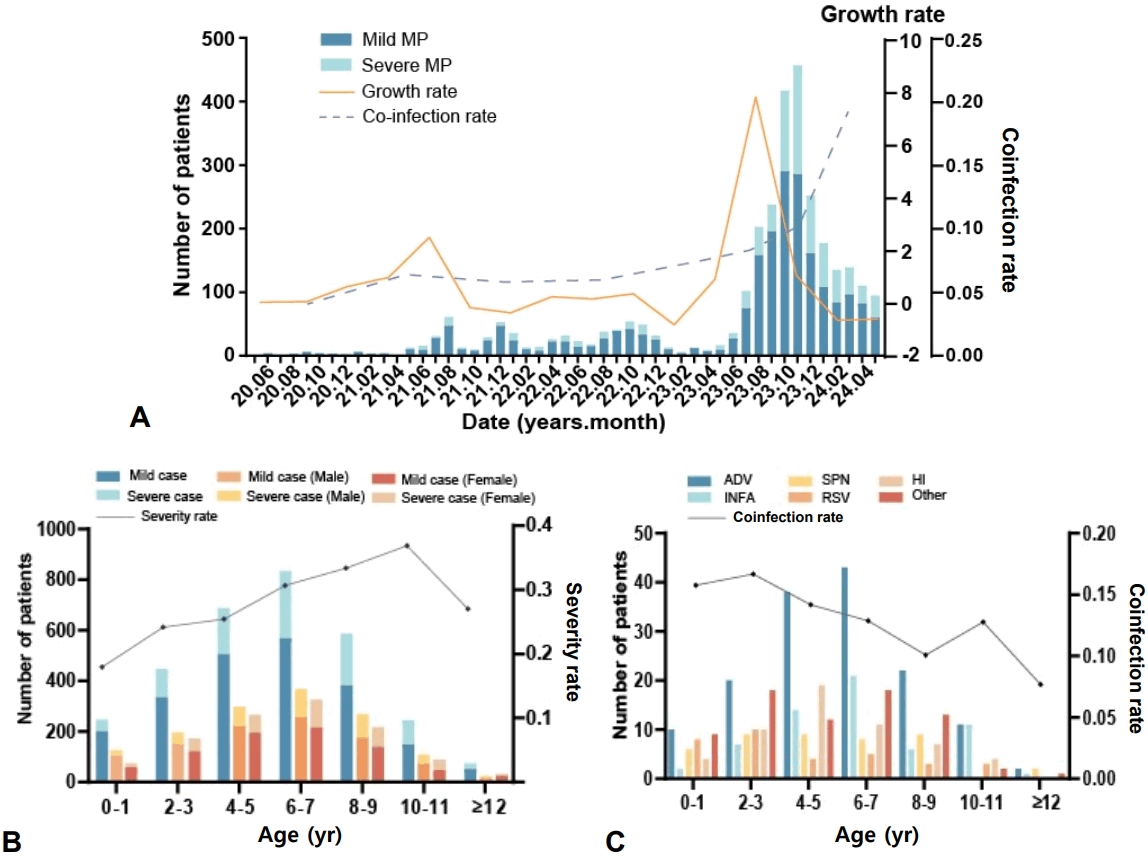
Among the 3,060 pediatric patients, 86.86% were identified with a simple MP infection. The remaining 13.14% showed codetection of MP along with nucleic acids from other pathogens. Among these coinfected cases, 11.73% involved 2 pathogens and 1.41% involved 3 pathogens (Fig. 2A–C). In the context of triple-pathogen coinfections, the most prevalent combination identified was MP, Streptococcus pneumoniae, and Haemophilus influenzae, which accounted for 27.9% of such cases (Fig. 2A). In the context of dual-pathogen coinfections, the most frequently observed coinfection was MP with adenovirus, identified in 138 cases. This was followed by MP coinfection with influenza A virus, which occurred in 69 cases, and MP alongside Haemophilus influenzae, noted in 39 instances (Fig. 2B). Overall, among all coinfected children, adenovirus was the most prevalent additional pathogen (Fig. 2D).
Coinfection patterns among children with MPP in Henan Province, China, from June 2020 to May 2024. (A) Coinfection involving 3 pathogens. (B) Coinfection involving 2 pathogens. (C) Proportion of coinfection cases. (D) Number of coinfection cases categorized by pathogen combinations. MPP, Mycoplasma pneumoniae pneumonia; MP, Mycoplasma pneumoniae; ADV, adenovirus; IAV, influenza A virus; HI, Haemophilus influenzae; SPN, Streptococcus pneumoniae; RSV, respiratory syncytial virus; HPIV, human parainfluenza virus; OC, oral candidiasis; Rhv, rhinovirus; EBV, Epstein-Barr virus.
3. Representativeness of the genotyped population
To assess the representativeness of the subcohort selected for in-depth genotypic analysis, we compared the 137 genotyped patients with the total hospitalized MPP population (n=3,060). As detailed in Supplementary Table 1, the 2 groups were well-matched in terms of basic demographics and temporal distribution. There were no statistically significant differences in age, sex ratio, or the proportion hospitalized during the 2023–2024 outbreak period. This indicates that the genotyped subcohort was not biased toward older children or concentrated in the recent epidemic wave. Importantly, the genotyped group exhibited a significantly higher ratio of severe MPP (42.3% vs. 32.1%, P=0.012). This moderate enrichment for severe cases is consistent with the clinical indications for bronchoscopy (which targets more severe or complicated presentations) and provides a suitable case mix for investigating associations between bacterial lineage and disease severity.
4. Analysis of MP genotyping and its correlation with disease severity and antibiotic resistance loci
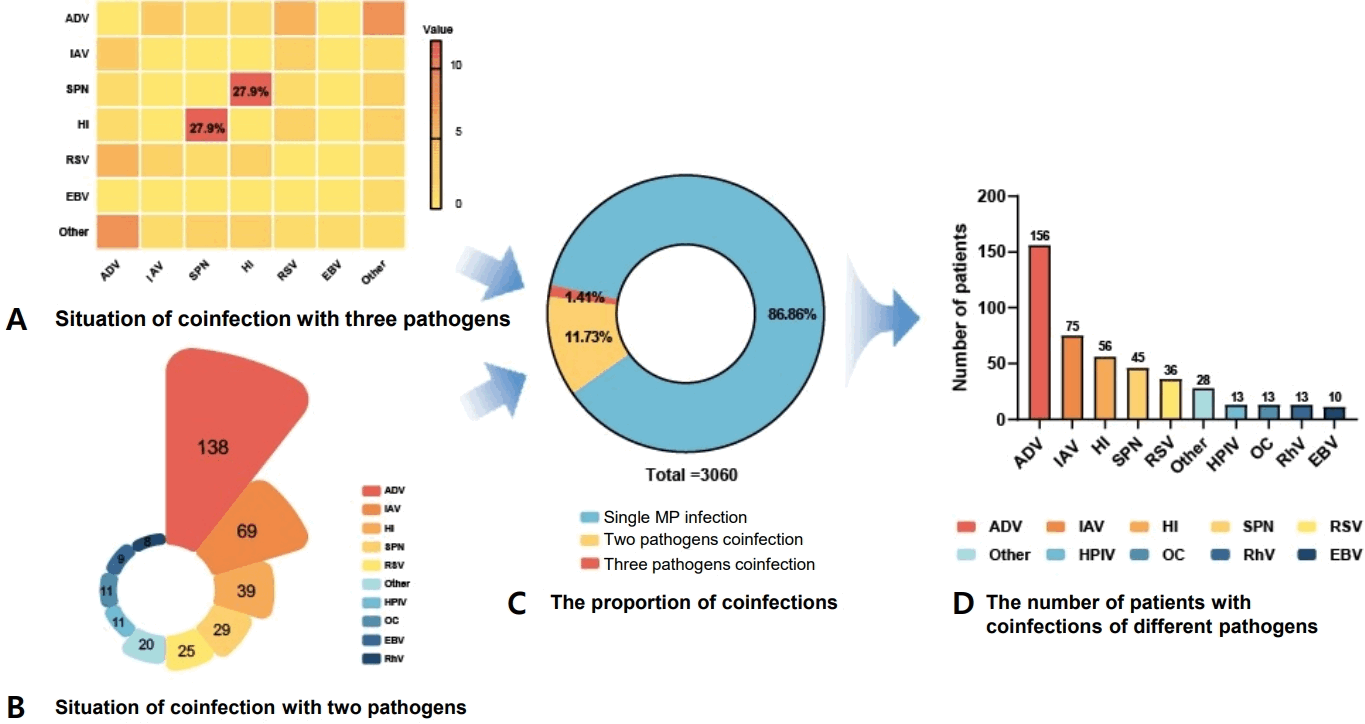
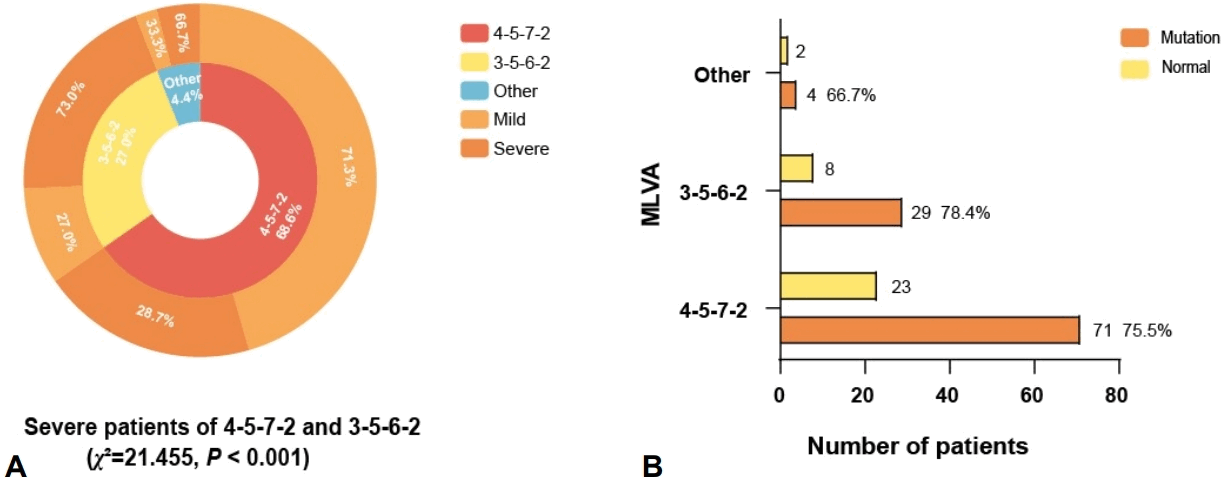
Genotyping was successfully performed on BALF samples from 137 pediatric patients. MLVA typing identified 2 predominant genotypes: the 4-5-7-2 genotype, which was the most prevalent (68.6%), and the 3-5-6-2 genotype (27.0 %). A striking disparity in disease severity was observed between these 2 types. The severe case rate associated with the 3-5-6-2 genotype was 73.0%, significantly higher than the 28.7% observed for the 4-5-7-2 genotype (χ2=21.455, P<0.001) (Fig. 3A). Furthermore, the analysis of macrolide-resistant MP mutations indicated that 75.9% of the children exhibited the A2063G mutation, which was the sole mutation site detected. The 3-5-6-2 genotype exhibited the highest mutation ratio at macrolide-resistant loci (78.4%), although this difference was not statistically significant across all genotypes (Fig. 3B).
Distribution of MLVA types among the 137 patients and their associations with disease severity and mutation at macrolide-resistant loci. (A) Proportion of severe cases by MLVA type. (B) Number and proportion of mutations at macrolideresistant loci among isolates by MLVA type. MLVA, multilocus variable-number tandem-repeat analysis.
In the MLST analysis, the ST3 genotype emerged as the most prevalent, identified in 57.7% of all patients, and was associated with a severe case ratio of 31.6%. The second most common genotype, ST14, accounted for 26.3% of samples and showed the highest severity ratio among all types at 72.2% (Fig. 4A). The difference in severity distribution between these 2 major genotypes was statistically significant (χ2=16.489, P<0.001). This distribution pattern aligns with the findings derived from MLVA typing. Furthermore, the macrolide-resistant loci mutation ratio in ST14 strains was 80.6%, which is comparable to that observed in the MLVA genotype 3-5-6-2. The mutation ratios among the remaining genotypes remained relatively stable, with an average of approximately 70% (Fig. 4B).
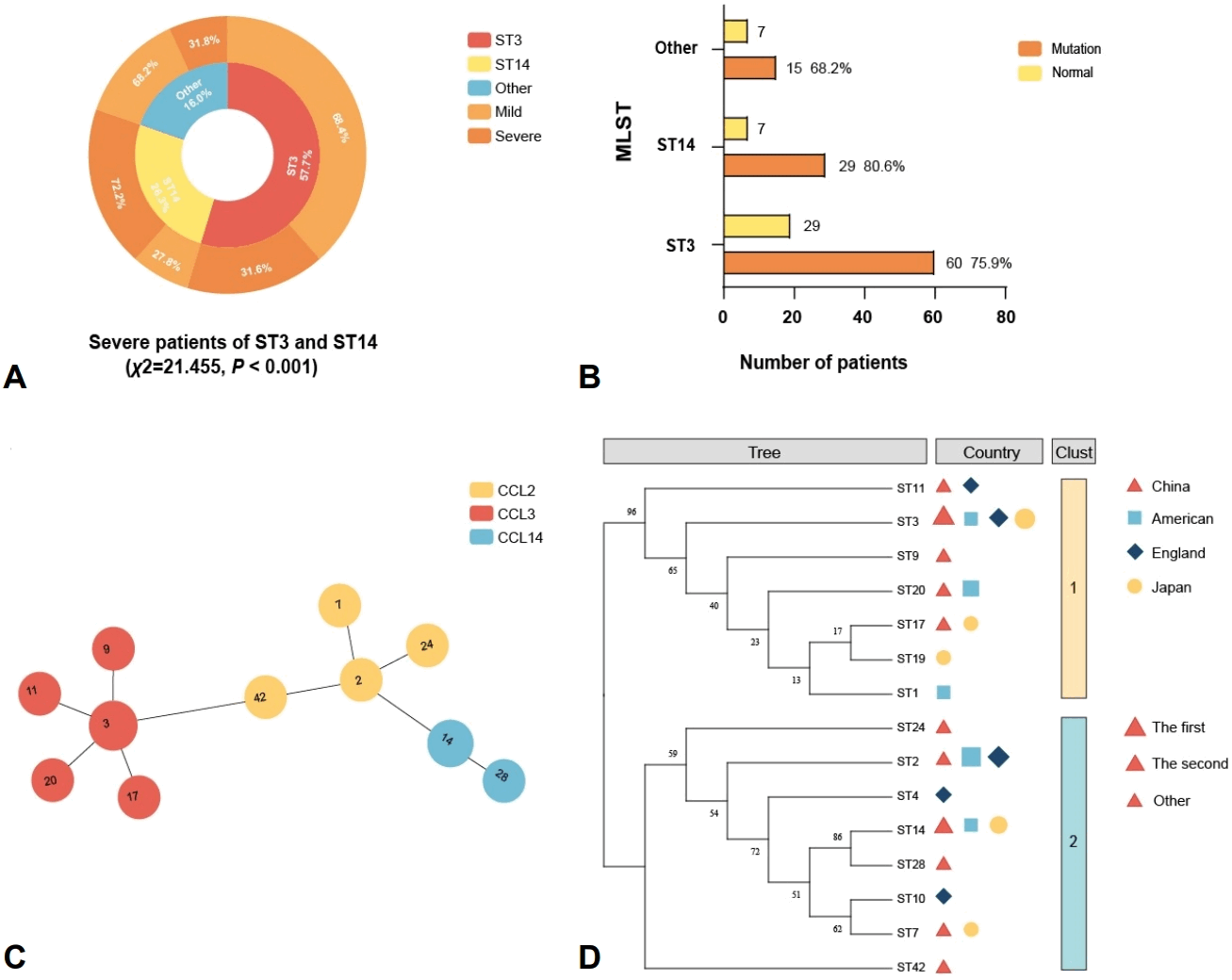
Distribution of MLST types and their association with disease severity, mutation at macrolide-resistant loci, and genetic relatedness among the 137 patients. (A) Proportion of severe cases by MLST type. (B) Number and proportion of mutations at macrolide-resistant loci isolates by MLST type. (C) Minimum spanning tree analysis illustrating genetic relationships among MLST types. (D) Phylogenetic tree analysis of geographical distribution of predominant MLST genotypes. CCL, common cluster label; MLST, multilocus sequence typing.
5. Phylogenetic clustering validates a hypervirulent lineage associated with severe disease
To investigate whether the observed difference in virulence between ST14 and ST3 extends to their broader phylogenetic lineages, we performed an MST analysis followed by CCL assignment. The findings showed that all MP strains were categorized into 3 distinct clusters. Among these, ST3, ST11, ST17, and ST20 were classified within CCL3 (n=97), whereas ST14 and ST28 constituted CCL14 (n=35) (Fig. 4C). The chi-square test results indicated that strains belonging to the CCL14 cluster were significantly more likely to cause severe MPP in patients compared to those in the CCL3 cluster (χ2=19.39, P<0.001). This robust association confirmed that the heightened virulence was a characteristic of the broader CCL14 lineage.
To determine whether the association between the CCL14 lineage and severe disease was independent of other factors, we performed a multivariable logistic regression analysis (Supplementary Table 2). After adjusting for age, sex, sample collection time, macrolide resistance mutation, infection with the CCL14 lineage remained the strongest and statistically significant independent risk factor for severe MPP (aOR, 5.60; 95% CI, 2.48-12.68; P<0.001). Age, sex, sample collection time, macrolide resistance mutation were not independently associated with severity in this model. The Hosmer-Lemeshow test indicated a good fit of the model (P=0.79).
Consequently, subsequent comparisons of clinical indicators focused directly on the differences between these 2 strain clusters. Prior to comparison, the normality of these laboratory parameters was assessed using the Shapiro-Wilk test. Significant differences (P<0.05) were observed between CCL3 and CCL14 in three clinical parameters: platelet count (t=2.21, P=0.032), D-dimer level (U=977.00, P=0.001), and complement C4 concentration (t=2.21, P=0.029). These differences suggest a potential association with enhanced inflammatory responses and changes in coagulation function among patients. Furthermore, no statistically significant difference was observed in the proportion of different strains before and after the outbreak in 2023 (χ2=0.48, P=0.490). For further clinical characteristics, refer to Table 1.
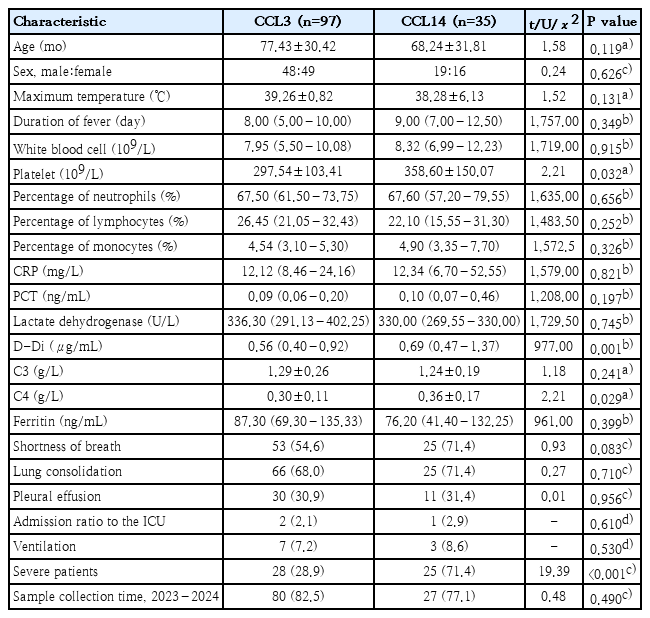
Correlation analysis of MLST typing and clinical indicators
6. Geographical distribution of predominant genotypes
Given the clinical significance of the hypervirulent CCL14 (ST14) lineage, we conducted a geographical distribution analysis to assess the prevalence of predominant strains in other regions. We integrated global MLST profiles to construct a phylogenetic tree for comparative analysis. The results indicated that ST3 and ST14 belong to distinct genotype clusters and are predominantly prevalent in Chinese and Japanese populations; ST2 and ST20 exhibit a relatively higher prevalence in the American population, whereas ST2 and ST3 are the predominant sequence types in the UK (Fig. 4D). This indicates that there are significant regional differences in the distribution of different MP strains, and confirms the predominance of the ST14 strain in Asia. Consequently, for patients in Asian regions, greater clinical vigilance should be exercised regarding the potential progression of the disease to severe forms.
Discussion
This study, based on epidemiological surveillance of pediatric MPP in central China from 2020 to 2024, revealed a significant resurgence of MP infections during the postpandemic period, characterized by a sharp increase in cases in 2023 and an elevated incidence of coinfections. In light of this context, we have identified the ST14/3-5-6-2 genotype and its associated CCL14 lineage as significant risk factors contributing to the development of severe MPP in this region. This finding establishes a molecular epidemiological foundation for a comprehensive understanding of the current severe clinical landscape of MPP.
Following the stabilization of the COVID-19 pandemic, MP experienced a large-scale local outbreak in autumn 2023, accompanied by a corresponding increase in coinfection ratios. Among the co-occurring pathogens, adenovirus was the most prevalent. On one hand, this indicates that changes in herd immunity levels may have contributed to the increased transmission of MP. On the other hand, it suggests that coinfection could further exacerbate the clinical burden associated with the disease [21,22]. Furthermore, our genotyping results indicate that there was no statistically significant alteration in the proportion of the main MP genotypes before and after the MP epidemic in 2023. This suggests that the current MP epidemic was not attributable to genotype changes. Likewise, Yan et al. [23] also reported that the 2023 resurgence in China is a continuation of the pre-COVID epidemic, rather than emergence of novel variants.
There is currently no consensus regarding the association between MP genotypes and clinical disease severity. While some research has suggested a potential correlation between specific genotypes and both clinical disease severity [16] and levels of inflammatory biomarkers [24], other studies have reported no significant association between MP typing and clinical outcomes [25,26]. This discrepancy may stem from critical methodological variations, including heterogeneous study populations and differing severity definitions. A pivotal factor, which our study design specifically addresses, is the source of clinical specimens. As a lower respiratory tract infection, the causative pathogen in MPP is most accurately reflected in BALF. The use of upper respiratory tract specimens (e.g., pharyngeal swabs), common in many studies, likely introduces a dilution effect by including colonizing strains or cases of mild infection, thereby obscuring the true relationship between virulent genotypes and the severe inflammatory response in the lungs. Our consistent findings from both MLVA and MLST, based exclusively on BALF samples, provide robust evidence that resolves these inconsistencies and firmly establishes the ST14/3-5-6-2 genotype as a high-risk variant.
Although P1 typing was not performed, our identification of the hypervirulent CCL14 lineage (ST14/3-5-6-2) aligns with established molecular epidemiology, as this genotype corresponds to P1 subtype 2 [20,25]. This is highly relevant, as P1 subtype 2 strains have been extensively reported to be closely linked to severe illness and robust inflammatory responses across multiple regions globally [27,28]. Thus, our data provide genotypic evidence that the clinical severity associated with P1 subtype 2 may be driven by the expansion of specific, hypervirulent clonal complexes like CCL14. Future studies should employ comparative or whole-genome sequencing to pinpoint the specific virulence determinants that are enriched in these lineages, moving beyond association to elucidate the precise molecular mechanisms driving severe disease.
Additionally, our genotyping results reveal a phenomenon that requires attention: the divergence between epidemic dominance and pathogenic dominance. While ST3/4-5-7-2 is the predominant strain in terms of prevalence, ST14/3-5-6-2 (CCL14) emerges as the primary contributor to severe clinical outcomes. This phenomenon indicates that in the epidemiological surveillance of MP, merely monitoring the number of cases and the proportion of strain composition is insufficient. It is imperative to concurrently assess changes in the virulence of dominant strains.
To examine the locally prevalent strains within a global context, we conducted a comprehensive analysis of their geographical distribution. Our findings indicate that ST3 and ST14 are the predominant genotypes in Asian countries (China and Japan), whereas genotypes including ST2 and ST20 are more prevalent in European and American regions. This distribution pattern aligns with findings from previous studies [9,29,30], providing strong evidence that the CCL14 lineage constitutes a high-risk clone widely circulating in Asia.
The simultaneous elevation of complement C4, D-dimer, and platelet count in patients infected with the CCL14 lineage reveals severe immune dysregulation. We hypothesize that infection with the CCL14 lineage triggers an exaggerated activation of the classical complement pathway, as evidenced by elevated C4 [31]. This intense inflammatory cascade, in turn, leads to endothelial injury and a consequent prothrombotic state, manifesting as elevated D-dimer. The significant increase in platelet count may represent a reactive thrombocytosis to the severe inflammation, or potentially a more direct interaction between the pathogen and the host's coagulation system [32-34]. This triad of complement activation, hypercoagulability, and intense inflammation likely constitutes the core pathological mechanism underpinning the severity of CCL14-associated MPP.
The interpretations of this study are subject to several limitations. It is crucial to note that our genotypic and subsequent analyses were performed on a cohort defined by clinical need for bronchoscopy. This selection criterion inherently enriches for moderate-to-severe cases, which, while providing a powerful dataset to investigate drivers of severe disease, limits the generalizability of our prevalence estimates to the broader, milder MPP spectrum. Second, our study cannot assess the relative virulence of the CCL14 lineage in causing initial or mild community-acquired infection. Therefore, our findings clearly demonstrate that the CCL14 lineage is associated with severe MPP specifically within the clinically consequential patient population that presents with or progresses to more severe disease necessitating invasive sampling.
In conclusion, our study elucidates the evolving epidemiological landscape of MP in the post-COVID-19 pandemic era and identifies the ST14/3-5-6-2 (CCL14) lineage as a predominant driver of severe disease in central China, characterized by enhanced inflammatory potential.
Supplementary materials
Supplementary Tables 1-2 are available at https://doi.org/10.3345/cep.2025.02796.
Comparison between the genotyped patients and the hospitalized MPP patients
Multivariable logistic regression analysis of factors associated with severe MPP
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This work was supported by China Postdoctoral Science Foundation (No. 2024T170246, No. 2024M750815), supported by the Open Grant from the Pingyuan Laboratory (No. 2023PY-OP-0202), supported by the Open Project of Key Laboratory of Infection and Immunity of Anhui Higher Education Institutes (No. I&I-2024-R01), supported by the Open Project of State Key Laboratory for Zoonotic Diseases, supported by Program for Innovative Talents in Higher Education Institutions of Henan Province (No. 25HASTIT055), and supported by Outstanding Youth Science Foundation of Henan Province (No. 252300421122).
Acknowledgments
We sincerely thank the Biobank of Henan Children’s Hospital for their help with the sample storage.
Author contribution
Conceptualization: ZL, YJ; Data curation: CH, SW, GJ, YY; Formal analysis: TS, YW; Funding acquisition: YJ; Methodology: FW, SC; Project administration: GD, YT; Visualization: GW, CH; Writing - originaldraft: ZL, CH; Writing - review&editing: YJ, GD