A case of Becker muscular dystrophy with early manifestation of cardiomyopathy
Article information
Abstract
An 18-year-old boy was admitted with chest discomfort, nausea, and dyspnea at rest. At the age of 3 years, he underwent muscle biopsy and dystrophin gene analysis owing to an enlarged calf muscle and elevated serum kinase level (6,378 U/L) without overt weakness; based on the results, Becker muscular dystrophy (BMD) was diagnosed. The dystrophin gene showed deletion of exons 45 to 49. He remained ambulant and could step upstairs without significant difficulties. A chest roentgenogram showed cardiomegaly (cardiothoracic ratio, 54%), and his electrocardiogram (ECG) showed abnormal ST-T wave, biatrial enlargement, and left ventricular hypertrophy. The 2-dimensional and M-mode ECGs showed a severely dilated left ventricular cavity with diffuse hypokinesis. The systolic indices were reduced, including fractional shortening (9%) and ejection fraction (19%). Despite receiving intensive medical treatment, he died from congestive heart failure 5 months after the initial cardiac symptoms. We report a case of BMD with early-onset dilated cardiomyopathy associated with deletion of exons 45 to 49. Early cardiomyopathy can occur in BMD patients with certain genotypes; therefore, careful follow-up is required even in patients with mild phenotypes of BMD.
Introduction
Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD) are allelic disorders of the dystrophin gene at the Xp21 locus1,2). Dystrophin plays an essential structural role in both cardiac and skeletal muscle, protecting the sarcolemma from mechanical stresses of muscle contraction3). Whereas DMD is characterized by the total absence or dysfunction of the dystrophin protein, BMD is characterized by reduced expression of the protein4). While respiratory failure is the main cause of mortality (mostly in the third decade of life) in DMD4), heart failure is the primary cause of death in BMD5). In addition, the severity and the age of onset of cardiac involvement in BMD shows no correlation to skeletal muscle involvement. According to the previous reports, cardiac involvement in BMD patients is detectable at later ages with an average onset age of 28.7±7.1 years6), and advanced cardiomyopathy is rare in BMD patients below 20 years5). Moreover, few cases of severe cardiomyopathy in BMD have been reported to date5,6).
Herein, we report the case of a patient with BMD who presented with dilated cardiomyopathy (DCMP) at the age of 18 years, and we discuss the factors that may influence early manifestation of DCMP in BMD patients with a review of the literature.
Case report
An 18-year-old boy was admitted to Seoul National University Children's Hospital; he experienced chest discomfort, nausea and dyspnea at rest. The patient, with no known family history of significant muscle disease, was first examined at 3 years of age because of enlarged calves. At the time of the first examination, blood tests revealed elevated serum levels of creatine kinase (6,378 U/L). The electromyogram showed myopathic changes, consisting of small polyphasic potentials. The biceps muscle biopsy revealed dystrophic features. Analysis of the dystrophin-encoding gene by multiplex polymerase chain reaction showed a deletion of exons 45 to 49. The patient remained ambulant with mild proximal weakness and has been able to ascend stairs without holding the lap one by one at the age of 18 years. At the time of admission his blood pressure was 118/61 mmHg and his pulse rate was 105 per minute without irregularity, and no evident heart murmur or rale was audible. The manual muscle test revealed only minimal weakness (4/5 or more) of the lower proximal muscles, and the patient had no subjective difficulties when standing from the squatting position. Chest roentgenogram showed cardiomegaly (cardiothoracic ratio=54%), and an electrocardiogram (ECG) exhibited an abnormal ST-T wave, biatrial enlargement, and left ventricular hypertrophy (Fig. 1). Markers of inflammation and viral titer were absent. Two-dimensional and M-mode echocardiograms showed a severely dilated left ventricular cavity with diffuse hypokinesis (Fig. 2). Endocardial systolic motion was reduced. Left ventricular wall thickness was normal. Systolic indices were reduced, including fractional shortening (9%) and ejection fraction (19%). The patient's left ventricular function deteriorated gradually in spite of intensive medical treatment for heart failure, and he died from congestive heart failure 5 months after initial cardiac symptoms at the age of 18 years and 4 months.
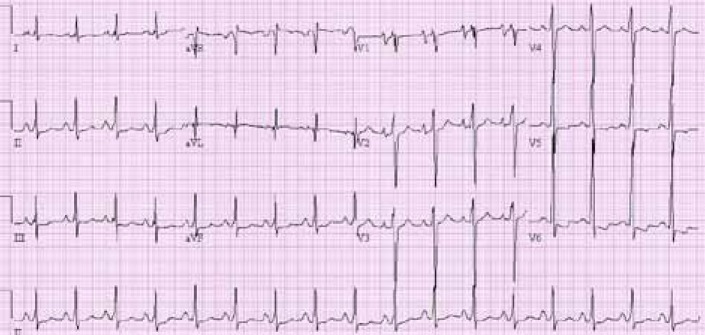
Electrocardiogram showing abnormal ST-T wave, biatrial enlargement, and left ventricular hypertrophy.
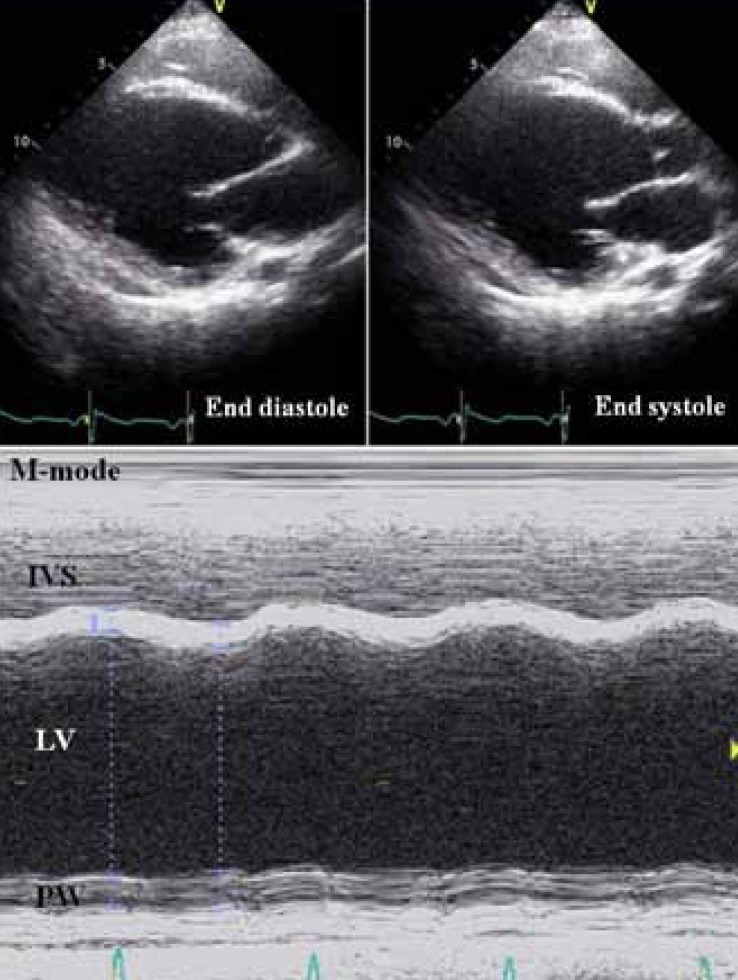
The upper panel showing the left ventricular long-axis view in the 2-dimensional echocardiograms, and the lower panel is the left ventricle in an M-mode echocardiogram. Both the panels exhibit a dilated left ventricular cavity with severely reduced contraction (end-diastolic left ventricular dimension, 73 mm; ejection fraction, 19.1% by the Teichholz equation). IVS, interventricular septum; LV, left ventricular cavity; PW, left ventricular posterior wall.
Discussion
We report a case in which the patient had a dystrophin gene deletion, of exons 45 to 49, and died from early-onset DCMP with BMD at the age of 18 years. Cases of early onset of cardiac failure are rare although there is evidence of frequent progressive cardiac involvement in BMD, characterized by the development of DCMP7,8).
In BMD, asymptomatic cardiac involvement develops in over 70% of cases, and these patients remain asymptomatic for a longer period9). Only up to one-third of BMD patients develop DCMP with symptomatic heart failure10,11). Cardiac involvement of BMD was reported to be uncommon in patients under 16 years of age, whereas more than 70% of patients demonstrated pathologic cardiac findings by the age of 40 years12,13). The median age of onset of fractional shortening less than 25% was reported to be 30 years14).
Several reports that cardiac dysfunction is not clearly correlated with skeletal muscle severities in DMD/BMD patients have been described, and they suggested possible pathogenesis and genotypic correlations. However, the association of genotype with DCMP has not been clearly established.
Jefferies et al.15) revealed an association between mutations involving exons 12 and 14 to 17 or 31 to 42, and early onset of DCMP. In addition, mutations involving exons 51 or 52 appeared to decrease the risk of cardiac involvement; however, because of the relatively short follow-up period, this could not be established with certainty. Yoshida et al.16) also suggested that a deletion around exon 1 may severely damage the expression and/or function of dystrophin selectively in cardiac muscle, but not in skeletal muscle. Involving exon 13 might also be associated with cardiac involvement in DMD/BMD17). Ramelli et al.18) reported the isolated deletion of exon 48 seems to be correlated with a mild course of the disease. Magri et al.19) reported that BMD patients with duplications of dystrophin gene experienced milder cardiac and respiratory involvement than patients with deletions, as demonstrated by the later age of onset.
Kaspa et al.3) suggested reasonable correlations between specific regions of the dystrophin gene and the age of DCMP onset in BMD patients through analysis of a large series of dystrophinopathy patients. BMD patients with deletion mutations affecting any portion of the actin-binding amino-terminal domain of dystrophin (exons 2 to 9) or exons 45 to 49 are at risk of developing DCM in their second and third decades of life, respectively3).
They also provided novel evidence of a strong association of DCMP with specific structural alterations of the dystrophin rod domain: DCMP with affecting exons 45 to 49 is more sensitive to altered phasing of the spectrin repeats rather than absence or presence of any individual exons. They suggested that the alterations caused by out-of-phase mutations extend beyond the spectrin repeats unit and may lead to a severely altered configuration of the rod domain, ultimately affecting the entire dystrophin (skeletal and cardiac) protein. In this case, the patient had a deletion affecting exons 45 to 49, encoding spectrin repeats 17 to 19, that preserves hinge 3 of the dystrophin protein showed later onset and slowly progressive skeletal muscle symptoms instead of earliest onset DCMP. This is in agreement with studies on dystrophin-null mdx mice expressing a mini-dystrophin construct that lacks the 45 to 49 region but has an intact hinge 3 domain. In these mice, only a partial restoration of cardiac function was achieved despite complete rescue of the skeletal muscle pathology20), which also suggested that cardiac dystrophin may be particularly sensitive to structural disruptions of exons 45 to 49 compared with skeletal muscle dystrophin.
Thus, we can assume that the early-onset DCMP and advanced heart failure in our case are related to the deletion site of exons 45 to 49 in the dystrophin gene, which is a more sensitive region in cardiac muscle dystrophin compared to that of skeletal muscle. To clarify the genotype-phenotype correlations of cardiac function in dystrophinopathy, further longitudinal large-scale epidemiologic study with functional or tissue-specific dystrophin expression studies of skeletal muscle dysfunction and cardiac symptoms might be necessary. Early cardiac evaluation and aggressive noninvasive imaging of young patients with muscular dystrophy can help identify those patients who are most likely to benefit from heart failure therapy15). On the basis of the results of previous studies, all the subjects with BMD who underwent drug therapy (a combination of digoxin and angiotensin-converting enzyme inhibitors) ultimately achieved normalization of the left ventricular dimension and systolic function compared with the DMD group, which did not show as dramatic an improvement15). Bushby et al.21) also recommended that BMD patients should undergo cardiac evaluation (ECG and echocardiogram) at diagnosis and be screened for the development of cardiomyopathy at least every 5 years. Considering that skeletal muscle weakness is not correlated with progression of cardiomyopathy in some of the BMD patients, alert and regular cardiac functional evaluation is necessary even in patients with mild and slowly progressive proximal weakness, particularly in cases with deletions affecting exons 45 to 49 as in our case, and patients with dystrophin mutations involving the amino terminal domains.
In conclusion, we report here a BMD case with dystrophin deletion from exon 45 to 49, who presented with early cardiomyopathy, and we also recommend careful and regular cardiac evaluation of BMD patients who have mutations affecting sensitive sites of dystrophin, to monitor cardiac involvement.