Asymptomatic moyamoya syndrome, atlantoaxial subluxation and basal ganglia calcification in a child with Down syndrome
Article information
Abstract
Down syndrome, the most common chromosomal abnormality, may be associated with various neurologic complications such as moyamoya syndrome, cervical spinal cord compression due to atlantoaxial subluxation, and basal ganglia damage, as well as epileptic seizures and stroke. Many cases of Down syndrome accompanied by isolated neurologic manifestations have been reported in children; however, Down syndrome with multiple neurologic conditions is rare. Here, we have reported a case of Down syndrome in a 10-year-old girl who presented with asymptomatic moyamoya syndrome, atlantoaxial subluxation with spinal cord compression, and basal ganglia calcification. To the best of our knowledge, this is the first report of Down syndrome, in a child, which was accompanied by these 3 neurologic complications simultaneously. As seen in this case, patients with Down syndrome may have neurologic conditions without any obvious neurologic symptoms; hence, patients with Down syndrome should be carefully examined for the presence of neurologic conditions.
Introduction
The patient with Down syndrome may present with diverse neurologic manifestations, including moyamoya syndrome, cervical spinal cord compression related to atlantoaxial subluxation, and basal ganglia damage1-4). Although there are many cases of children with Down syndrome accompanied by isolated aforementioned neurologic manifestations, it is rare that children with Down syndrome have multiple neurologic problems, simultaneously. We describe a case of a child with Down syndrome, who had asymptomatic Moyamoya syndrome, atlantoaxial subluxation, and basal ganglia calcification, simultaneously.
Case report
A 10-year-old girl with Down syndrome was referred to the pediatric department from the ophthalmologic department of Ulsan University Hospital because both basal ganglia calcification was incidentally detected on a brain computed tomography (Fig. 1). She had been managed at the ophthalmologic clinic because of strabismus. Brain computed tomography (CT) was performed to identify whether her ophthalmologic manifestations, including esotropia of both eyes, amblyopia in the left eye, cyclotorsion of both eyes, and horizontal nystagmus, would result from brain parenchymal lesion.
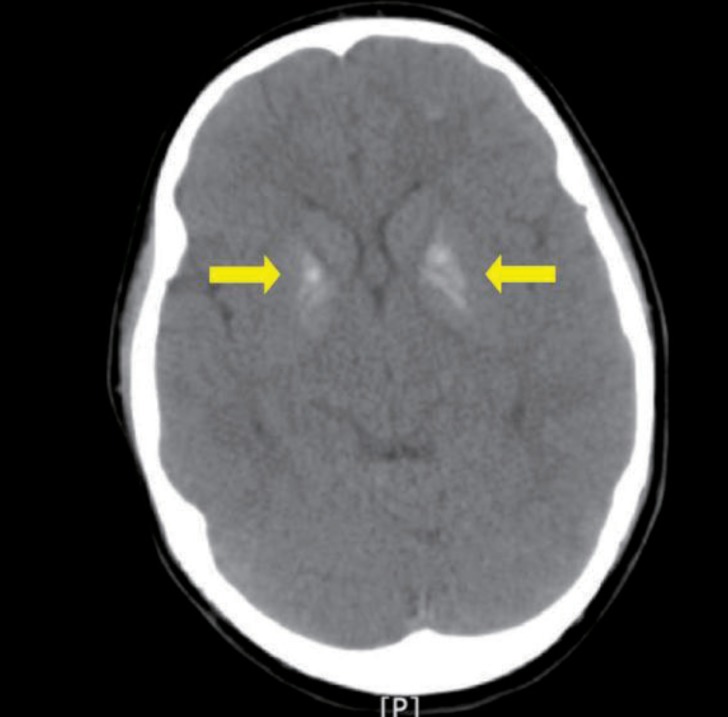
Brain computed tomography shows calcifications in the bilateral basal ganglia (arrow).
She was born at Ulsan University Hospital. At that time, she showed typical features of Down syndrome, and was diagnosed with Down syndrome by the identification of 47,XX,+21 on the cytogenetic study. Echocardiography after birth, which was performed due to cardiac murmur, revealed a 6.3 mm sized perimembranous ventricular septal defect and two small sized atrial septal defects. She had been examined by regular follow-up echocardiography evaluation without surgery, and was presented with no symptoms associated with cardiac anomalies until the time of admission. She had moderate mental retardation with full scale intelligence quotient score of 44. Her parents stated that they had never noticed apparent neurologic symptoms of the patient, including transient motor weakness before the admission.
On physical examination at the time of admission, she showed typical features of Down syndrome, including hypertelorism, depressed nasal bridge, low-set ears, epicanthal folds, protruded tongue, small chin, and simian creases on both hands. On cardiac auscultation, systolic murmur was heard. On neurologic examination, she was fully alert and conscious. She showed esotropia of both eyes, but, the limitation of both eye movements was absent. She exhibited horizontal nystagmus, without vertical nystagmus. No obvious other abnormalities were revealed on the cranial nerve examination. Motor weakness of four extremities was absent, although muscle tone was slightly hypotonic. The deep tendon reflexes on both knees were slightly exaggerated, and Babinski sign was absent in both. On laboratory findings, we observed total calcium, 9.8 mg/dL; ionized calcium, 4.60 mg/dL; parathyroid hormone, 44.94 pg/mL; ceruloplasmin, 22 mg/dL; serum copper, 96 µg/dL; thyroid-stimulating hormone, 1.20 mIU/L; and free thyroxine, 1.49 ng/dL. Investigations for autoimmune (antinuclear antibody, antidouble strand DNA antibody, anticardiolipin antibodies, antiphospholipid antibodies, and antineutrophilic cytoplasmic antibody), prothrombotic (protein C, S, Factor V Leiden mutation, and antithrombin III) and metabolic (serum amino acid and urine organic acid) disorders were within normal limits. Echocardiography performed, during the admission, revealed 3-mm sized perimembranous ventricular septal defect with small to moderate left to right shunt, but good ventricular function. Brain magnetic resonance imaging and magnetic resonance angiography, which were performed during the admission, showed as follows; multiple tiny ischemic lesions in both frontal white matters, compression of the cervical cord due to displaced odontoid process at the level of C1, and severe stenosis of the bilateral internal carotid arteries with collaterals to the bilateral cerebral hemispheres (Figs. 2, 3A). Four vessels angiography revealed occlusion of the proximal portion of the right internal carotid artery, occlusion of the left internal carotid artery bifurcation and the left middle cerebral artery, transdural collaterals from the middle meningeal, the superficial temporal, and the occipital arteries, and hypertrophic right vertebral artery with prominent leptomeningeal collaterals to both cerebral hemispheres. Three dimensional cervical spine CT showed subluxation and rotation of C2 on C1 with spinal canal narrowing, and os odontoideum (Fig. 3B).
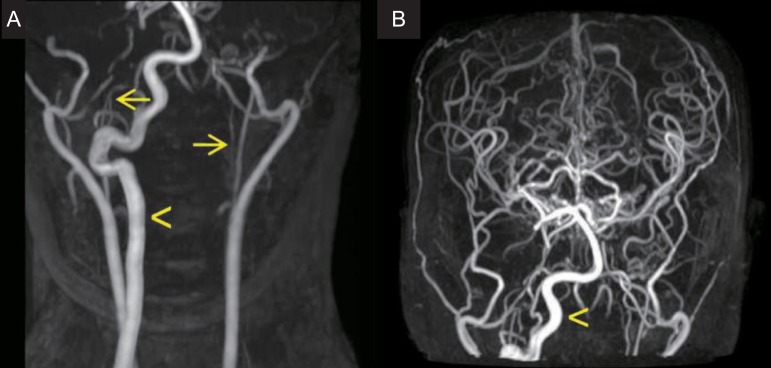
Time of flight magnetic resonance angiography. (A) Severe stenosis of the bilateral internal carotid arteries (arrows) is observed. (B) A hypertrophic right vertebral artery (arrowhead) supplies collateral blood flow to the bilateral cerebral hemispheres.
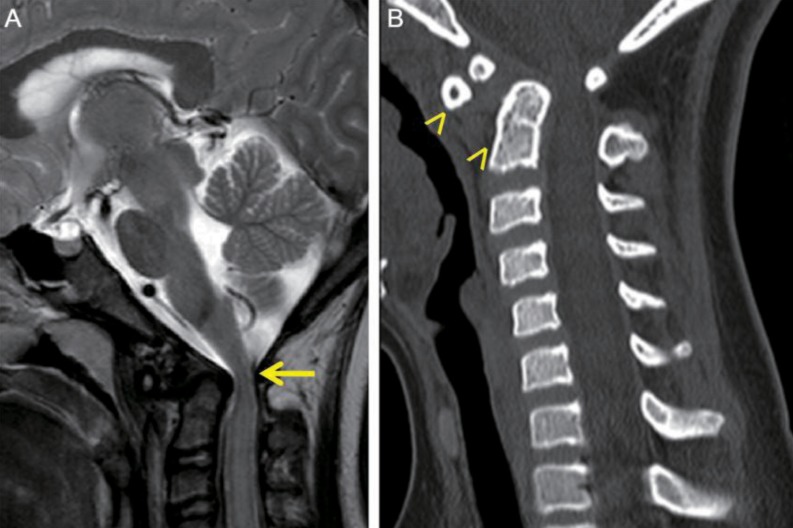
(A) T2-weighted sagittal image shows compression of the cervical cord (arrow), due to the displaced odontoid process at the level of C1. (B) Cervical spinal computed tomography scan shows subluxation of C2 with os odontoideum (arrowheads).
Her family moved to another province far from our hospital, several months after the evaluation. According to the telephone interview with the patient's mother, the patient had been doing well with no neurologic symptom and no special management during the 2 years since the admission to our hospital.
Discussion
Down syndrome may be associated with various neurologic complications, including moyamoya syndrome, cervical spinal cord compression related to atlantoaxial subluxation, and basal ganglia damage, as well as epileptic seizures, stroke, and Alzheimer disease1-4).
Moyamoya syndrome has been reported to occur with a higher frequency in Down syndrome than in the general pediatric population5). Moyamoya syndrome is a cerebrovascular disorder, characterized by a slowly progressive bilateral stenosis, eventual occlusion of the supraclinoid internal carotid arteries and produces collateral vessels. It is different from moyamoya disease in that moyamoya syndrome occurs secondary to a variety of slow progressive, occlusive cerebral vasculopathies and genetic syndromes, such as sickle cell disease, postradiation vasculopathy, Down syndrome, neurofibromatosis, and Williams syndrome2). The exact mechanism of vascular occlusion in Down syndrome with moyamoya syndrome is unknown, although several hypotheses have been proposed6-11). Patients with Down syndrome are generally predisposed to vascular abnormalities, such as abnormal nailbed capillary morphology, high pulmonary vascular resistance with congenital heart disease, abnormalities of retinal vessels and primary intimal fibroplasias6). In respect of the molecular basis, several proteins associated with the increased risk for vascular disease, such as alpha chains of collagen type VI, superoxide dismutase I, interferon gamma receptor and cystathionine beta synthase, are encoded on chromosome 216-9). Autoimmunity and protein C deficiency has also been postulated as other possible mechanisms associated with Down syndrome and moyamoya syndrome10,11). In our case, however, the work-ups for autoimmune and hypercoagulation disorders were all negative.
Atlantoaxial instability occurs in 10% to 30% of patients with Down syndrome, the majority of which are asymptomatic, with estimates of symptomatic disease ranging from 1% to 2%12). Atlantoaxial instability in the Down syndrome population may be secondary to bony anomalies, such as os odontoideum, which occurs in approximately 6% of children with Down syndrome13). Other proposed causes for atlantoaxial instability in Down syndrome include intrinsic collagen defects that lead to transverse ligament laxity, and a chronic inflammatory state that weakens the ligamentous structures14,15). Atlantoaxial subluxation narrows the anteroposterior diameter of the neural canal, potentially compressing the spinal cord at the level of C1. It is generally accepted that ligamentous instability at the atlantoaxial joint in patients with Down syndrome is unlikely to progress to clinically relevant subluxation12). However, those children with bony anomalies are at higher risk of progression and should be followed closely. In our case, although obvious neurologic manifestations associated with moyamoya syndrome and atlantoaxial subluxation did not appear, it should be kept in mind that neurologic symptoms may develop later, considering her age and the progressive characteristics of moyamoya syndrome and os odontoideum.
Incidence of basal ganglia calcification, in patients with Down syndrome, is very high compared to the general population (10-45% vs. 0.3-0.6%)16). Basal ganglia calcification in the normal population is occasionally observed in older patients above 40 years of age. In Down syndrome, basal ganglia calcification appears at a younger age, which perhaps indicates premature aging17). The pathogenesis of basal ganglia calcification in Down syndrome is unknown. Takashima and Becker16) found basal ganglia calcification was localized to a constant area of globus pallidus, calcification was increasingly severe with aging and calcification and amyloid degeneration of the adjacent blood vessels were present. They suggested that progressive structural change occurs in the vessel walls with advancing age in patients with Down syndrome. Basal ganglia calcification in Down syndrome is usually asymptomatic, and is detected incidentally by neuroimaging, although some reports associated with neurologic disorders, such as psychosis, movement disorder, have been documented18,19).
To the best of our knowledge, this is the first report that a child with Down syndrome had moyamoya syndrome, atlantoaxial subluxation, and basal ganglia calcification, simultaneously. Because patients with Down syndrome may have multiple neurologic problems simultaneously, even without obvious neurologic symptoms like our case, judicious practice will be necessary in the evaluations for patients with Down syndrome.
Notes
No potential conflict of interest relevant to this article was reported.