Congenital muscular dystrophy type 1A with residual merosin expression
Article information
Abstract
Congenital muscular dystrophy type 1A (MDC1A) is an autosomal recessive disorder characterized by hypotonia, elevated serum creatine kinase level, delayed motor milestones, white matter changes observed by brain magnetic resonance imaging, and normal intelligence. A mutation in the laminin α2 (LAMA2) gene, located at 6q22-23, is a genetic cause of MDC1A. Patients have merosin (laminin α2)-deficient skeletal muscles. However, the degree of merosin expression ranges from total absence to partial reduction. Patients with residual merosin expression have more variable and milder phenotypes than those with absolute merosin deficiency. We observed a Korean girl with MDC1A with residual merosin expression. Clinical presentation of this patient was typical except for late onset of the disease and external capsule involvement. Immunohistochemical staining of muscle fibers including merosin, is important to evaluate patients with hypotonia, delayed motor development, and abnormal white matter changes.
Introduction
Congenital muscular dystrophy type 1A (MDC1A) was first described in 1994 as a variant form of classic congenital muscular dystrophy (CMD)1). MDC1A is an autosomal recessive disorder, and patients show absent merosin (laminin α2) on skeletal muscle biopsy. A mutation in the laminin α2 (LAMA2) gene, located on 6q22-23, was later found to be a genetic cause of MDC1A2). Most patients with MDC1A show absent merosin in muscle immunohistochemistry studies, however, there have been some reports of cases of MDC1A with residual merosin expression3,4,5). MDC1A is very rare in Asian countries. Comparing with studies conducted in western countries2,3), only a few cases without merosin expression have been reported in the Asian population6,7,8). Here, we describe a patient with MDC1A and residual merosin expression.
Case report
The patient was born at 38 weeks of gestation with a birth weight of 3,500 gm. There was no history of decreased fetal movement and asphyxia at birth. She was a second child, with one 4-year-old brother with no medical problems. There was no history of parental consanguinity. She was healthy at birth, but her parents noticed that she had a delayed motor milestone at the age of 4 months as indicated by her inability to fully control her head. She was brought to Severance Children's Hospital for the first time at the age of 4 months and was hypotonic at that time. The traction test showed headlag, and the suspension test showed inverted U sign. Deep tendon reflexes were weakly preserved. She was fully examined for muscular disease at the age of 6 months, at which time she could not yet control her head. However the Bayley Scales of Infant Development for language and cognitive development were normal. Her serum creatine kinase (CK) level was elevated to 2,184 IU/L (0-50 norm) and her serum aldolase level was also high at 31.6 sigma U/mL (0-7.6 norm). In a nerve conduction study, both sensory and motor conduction were shown to be normal. Electrocardiography and echocardiography showed no abnormalities. Brain magnetic resonance imaging (MRI) performed at the age of 6 months showed excessive T2 hyper-intensity at the peritrigone and external capsule (Fig. 1). Repeated brain MRI at the age of 8 months showed progression of dysmyelination. We performed a muscle biopsy in the left vastus lateralis muscle. Hematoxylin and eosin staining showed marked muscle fiber size variation, increased endomysial fibrosis, many endomysial inflammatory cell infiltrations, and many necrotic and regenerative muscle fibers (Fig. 2). Modified Gomori trichrome staining did not show ragged red fibers, nemaline bodies, or rimmed vacuoles. Nicotinamide adenine dinucleotide-tetrazolium reductase staining showed disorganized inter-myofibrillar networks. These findings suggest muscular dystrophic changes. Immunohistochemical staining was performed using antibodies against the C-terminal of dystrophin, rod domain of dystrophin, N-terminal of dystrophin, α-sarcoglycan, β-sarcoglycan, γ-sarcoglycan, δ-sarcoglycan, dysferlin, α-dystroglycan, caveolin 3, and laminin α2. Staining against laminin α2 was partially decreased, while the others were normally expressed (Fig. 3). To confirm mutation in the LAMA2 gene, we performed direct sequencing of exons 14, 25, 26, and 27, all of which are known as common sites of mutation. However, no point mutations were identified.

Axial T2-weighted brain magnetic resonance images of the patient at 6 months of age shows high signal intensity in the white matter of the peritrigone and external capsule (A); slight progression of dysmyelination can be seen 2 months later (B).
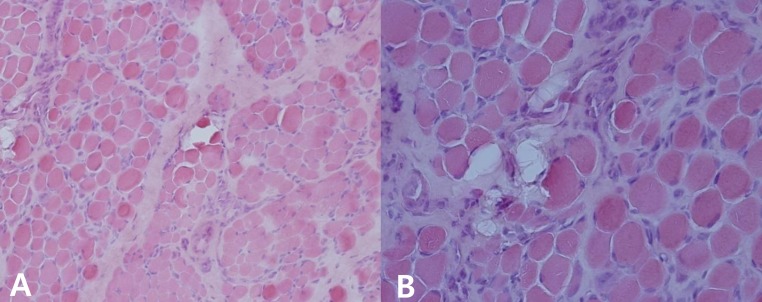
Hematoxylin and eosin staining shows marked muscle fiber size variation, increased endomysial fibrosis, numerous endomysial inflammatory cell infiltrations, and increased necrotic and regenerative muscle fibers (A, ×200; B, ×400).
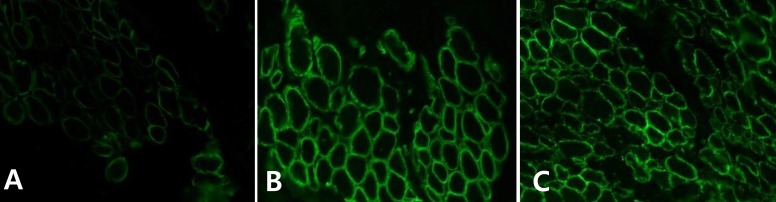
Immunohistochemical staining of the patient's muscle fiber demonstrates partially decreased laminin α2 expression (A, ×400) comparing with normal expression of dystrophin (B, ×400) and α-dystroglycan (C, ×400).
Discussion
Typical clinical features of MDC1A or merosin deficient congenital muscular dystrophy include severe floppiness at birth, elevated serum CK, delayed motor milestones, white matter changes as seen on brain MRI, and normal intelligence2). However, merosin-positive CMD generally presents with less severe clinical features and without any abnormalities on brain MRI2,5). Initially, MDC1A was thought to be a homogenous disease; however, case reports of atypical phenotypes and some cases showing only partially reduced merosin expression contributed to classifying the disease as a heterogeneous group3,5). There have been MDC1A patients who show late-onset weakness, mental retardation, seizures, subclinical cardiac involvement, or neuronal migration defects9). In particular, cases with residual merosin expression have more variable phenotypes than cases with absolute merosin deficiency. According to previous reports, patients with residual merosin tend to have milder phenotypes3,5). With regards to ambulation, a previous study showed that eight out of 12 patients with residual merosin achieved independent ambulation, whereas, out of four patients with absent merosin, none could walk independently5). Another study presented similar results: five out of 13 patients with residual merosin and two out of 33 patients with absent merosin could walk independently3).
In our case, the onset of symptoms was 4 months after birth, rather than during the neonatal period. Usually, merosin deficient patients present with symptoms within 7 days of birth, while patients with residual merosin have a later onset of symptoms. The other symptoms of this patient, such as hypotonia, normal intelligence, elevated serum CK, and the absence of seizures, are typical presentations of MDC1A patients. At present, she is 14 months old, and can sit without support but cannot get into a sitting position. We estimate that she has about a 50% probability of walking independently later in her life.
Brain MRI of this patient showed typical hyperintensity in the T2-weighted image. However, the patient's site of lesion is in the external capsule, which is an unusual area for this disease. A study of 25 Brazilian patients with MDC1A reported that bilateral white matter involvement in the parietal, frontal, and temporal regions was frequent, and brain stem, cerebellum, and internal and external capsules were also affected in a minority of cases10). However, there were no correlations with sites of white matter abnormalities and clinical status or merosin status.
To confirm mutation of the LAMA2 gene, we sequenced exons 14, 25, 26, and 27. This technique, while able to screen for point mutations in above 4 exons, is limited in its ability to detect large deletions or insertions or point mutations in the other sites. We did not further evaluate other possible mutations in the LAMA2 gene because of her parents' refusal. According to a previous report, a majority of mutations in the residual merosin group are splice site mutations, and frameshift mutations are less frequent3). This patient may have mutations in her LAMA2 gene, although we could not confirm the specific mutations because of incomplete study.
MDC1A is very rare in Asia, and the cases reported thus far are typically those without merosin, and have mostly been severe forms of MDC1A. In our study, the patient showed a typical phenotype of MDC1A, including features such as hypotonia, elevated serum CK, delayed motor development, and T2 hyperintensity on brain MRI. However, immunohistochemical staining of the muscle fibers showed partially decreased merosin levels and some atypical findings, such as a somewhat later age of disease onset and involvement of the external capsule. From our experience, performing immunohistochemical staining to assay the extent of merosin expression is an important procedure, and may be helpful in predicting the clinical course of the disease.
Acknowledgments
This work was supported by a National Research Foundation grant funded by the Korean government (MEST) (2010-0020353).
Notes
No potential conflict of interest relevant to this article was reported.