Multifocal kaposiform hemangioendothelioma of soft tissue with bilateral pulmonary involvement in an adolescent
Article information
Abstract
Kaposiform hemangioendothelioma (KHE) is a rare, locally aggressive vascular tumor of intermediate malignancy with resemblance to Kaposi sarcoma. It occurs predominantly in pediatric age groups as a cutaneous lesion with focal infiltration into the adjacent soft tissue and bone. Although visceral involvement is very uncommon, several cases with bone, retroperitoneal, or mediastinal involvement have been described. KHE has been reported to occasionally occur in unusual sites such as the thymus, tonsils, larynx, paranasal sinuses, deltoid muscle, spleen, uterine cervix, thoracic spine, and even the breast. Multifocal KHE is an extremely rare entity with few reports available in the literature, none of which describes pulmonary involvement. Herein, we report a unique case of multifocal KHE in a 13-year-old boy presenting with a huge soft tissue mass in the upper extremity complicated by bilateral pulmonary nodules that developed into large, necrotic tumor masses.
Introduction
Kaposiform hemangioendothelioma (KHE) is a rare distinctive vascular tumor of soft tissue that occurs almost exclusively in children. Original reports mainly emphasized on skin and retroperitoneum as the most common location of the tumor; however, Zukerberg et al.1) later described more varied presentations of KHE. Histologically, it is composed of infiltrating nodules of vessel forming slit-like, crescentic capillaries with individual lumens containing red blood cells. It grows rapidly with focal extension into the adjacent skin, soft tissue and bone1,2). In the extremities, it usually appears as a superficial or deep tissue mass3). There are various reports of single organ involvement of KHE in different locations and also a few studies discussing multifocal KHE. Multifocal KHE is an extremely rare entity with only occasional reports available in the literature. To the best of our knowledge, pulmonary involvement complicating KHE has not been previously described in English medical literature.
Case report
A 13-year-old boy was referred to the pediatric oncology clinic with a rapidly enlarging mass in left elbow for the past three months. Physical examination was unremarkable except for the mass in left elbow with extension to the arm and upper forearm causing complete limitation of the joint motion. The overlying skin had no discoloration. Complete blood counts and coagulation profile were all within normal limits. The radiographs demonstrated severe soft tissue swelling in elbow and periarticular tissues. Magnetic resonance imaging (MRI) of the left elbow revealed a large 11 cm×12 cm×16.5 cm heterogeneously enhancing cystic/solid circumferential mass with vague border in volar aspect of distal arm, antecubital fossa and proximal forearm, infiltrating the asssociated muscles, including brachialis, brachioradialis, extensor carpi radialis and all the muscles in the proximal forearm, as well as neurovascular bundles (Fig. 1A, B). The bone marrow signal intensity of the distal humerus, proximal radius and ulna were within normal limits with no apparent infiltration. The findings were most consistent with a soft tissue sarcoma. Chest computed tomography (CT) scan showed multiple variable sized pulmonary nodules predominantly in the lower zones of both lungs, ranging up to1.5 cm (Fig. 2A). Abdominopelvic CT scan was unremarkable. A whole body scan with 99mTc was normal and showed no evidence of involvement of the bones adjacent to the tumor. He underwent a wide local excision of the extremity tumor. Histologically, the tumor was composed of hyper- and hypocellular areas with a myxoid stroma. The tumor cells were predominantly composed of mildly pleomorphic cells with oval to spindle shaped vesicular nuclei and scant to moderate amount of cytoplasm. Many individual cells formed small lumens, containing red blood cells (Fig. 3A). Small- and medium-sized vessels were also present. Mitotic activity was infrequent in most of the tumor; however, a few glomeruloid nests composed of pleomorphic epitheliod cells with high mitotic activity were identified (Fig. 3B). The tumor was diagnosed as KHE with focal atypia and increased mitotis (glomeruloid epithelioid nests). Immuno histochemical study showed positivity of some tumor cells for CD31 (Fig. 4) and CD34. Accordingly, a combination of prednisone (starting dose of 2 mg/kg/day for a period of 1 month followed by gradual reduction within 3 months), propranolol and alpha-interferon (3 million units thrice weekly) was started. An obvious regression of the primary tumor resulted and interim chest CT scan after 3 months showed decrease in number of pulmonary nodules. However, he came back with high fever and severe respiratory distress 4 months later. Chest CT-scan at this time revealed increase in the size and number of the pulmonary nodules. Some of them had evolved into masses up to 6.2 cm with necrotic irregular centers and intense peripheral enhancement as well as bilateral pleural effusion (Fig. 2B, 2C). Aspiration of the pleural fluid showed no malignant cells on two occasions. Bilateral chest tubes were inserted and lung biopsy was taken by video assisted thoracoscopic surgery. Histologic findings of lung lesions were compatible with KHE. He also developed a hypodense 1 cm round lesion in the spleen in follow-up imagings that were undertaken in 3-month intervals. A repeat bone scan showed increased uptake of the radiotracer in multiple bones. Due to multifocal extension of the disease and regrowth of the primary tumor of the extremity the patient received bevacizumab (Avastin, Roche, Basel, Switzerland) (15 mg/kg every 3 weeks) and thalidomide (300 mg/day). Soft tissue tumor of the extremity and pulmonary involvement showed remarkable response after the first two courses of bevacizumab (Fig. 2D); nevertheless, shortly thereafter obvious regrowth of the primary tumor ensued. Likewise, progressive pulmonary involvement characterized by enlargement of bilateral huge tumoral masses and pleural effusion detected by chest x-ray and chest CT scan indicated unresponsiveness to the aforementioned therapies (Fig. 2E, F).
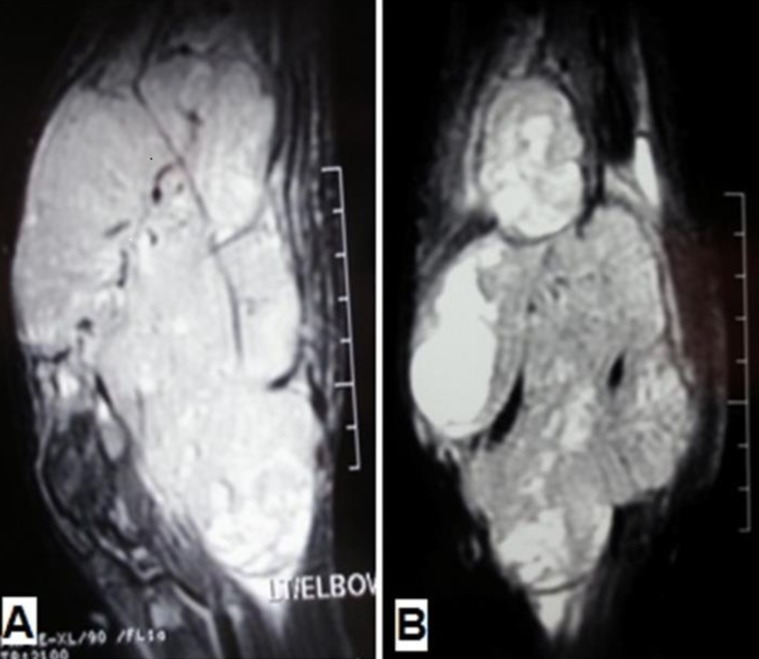
(A) Sagittal and (B) Coronal fat-saturated magnetic resonance images show a large heterogeneous mass in the antecubital fossa with extension to the distal arm and proximal forearm, infiltrating the corresponding muscles and encasing the neurovascular bundles.
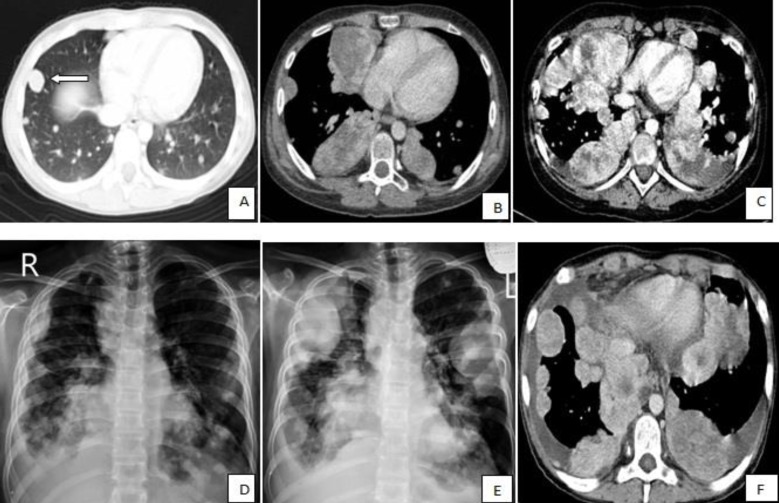
Thoracic computed tomography (CT) scans and chest radiographs show the changes in the size and number of multiple pulmonary nodules during the course of disease. (A) The CT scan taken at the time of diagnosis shows multiple, variable-sized pulmonary nodules with the largest nodule in the right middle lobe measuring 15 mm (white arrow); (B, C) The CT shows an increase in the size and number of multiple nodules and masses with central irregular necrosis and avid peripheral enhancement. Superimposed bilateral pleural effusion is also shown in panel C; (D) Chest radiograph shows a decrease in the size and number of nodules after administration of bevacizumab; and (E, F) Regrowth of the lesions and bilateral pleural effusion on chest radiograph and CT scan, respectively, indicate unresponsiveness of the patient to bevacizumab and thalidomide.
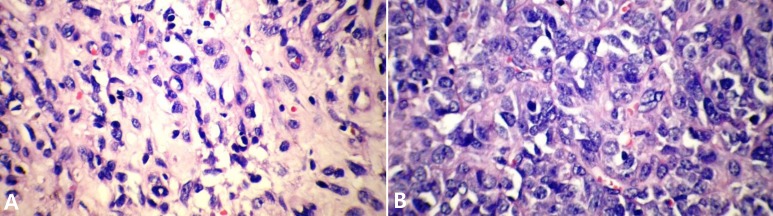
(A) Hypocellular area of tumor shows that individual cells form lumen that contain red blood cells (H&E, ×400). (B) Hypercellular area of tumor shows pleomorphic epithelioid cells and vascular space formation by tumor cells (H&E, ×400).
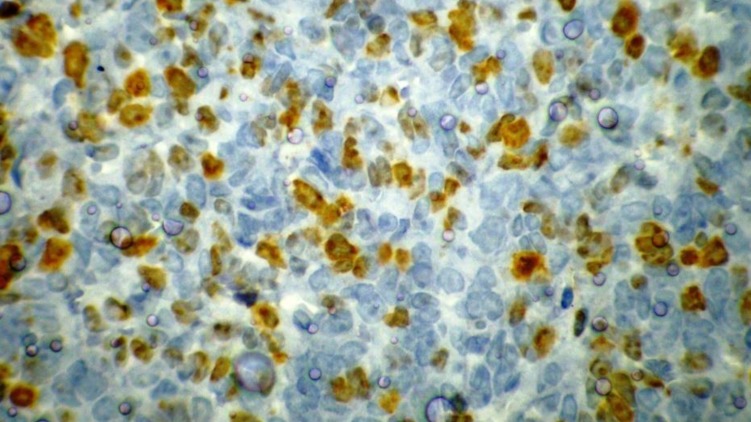
Immunohistochemistry indicates that some tumor cells are positive for CD31 (×400).
Discussion
KHE was first described in 1993 by Zukerberg et al.1) as a distinctive lesion of childhood with common features to both hemangiomas and Kaposi's sarcoma. It is a rare vascular tumor typically seen in children as a cutaneous lesion with ill defined borders. KHE is a locally aggressive spindled cell endothelial derived tumor of intermediate malignancy that may cross tissue planes from dermis into subcutis, fascia, muscle and the bone2). According to the largest recent series reported by Croteau et al.4) the prevalence of KHE is estimated to be as 0.91 case per 100,000 children. Over the past decade, there has been about one new case of KHE diagnosed in Massachusetts per year, yielding an incidence of 0.071 case per 100,000 children. In the first description of KHE by Zukerberg et al.1), 9 cases were reported, of which only two were beyond the first decade of life. Interestingly, Zukerberg et al.1) have also reported a case with a deep soft tissue mass of the arm with involvement of adjacent soft tissues, bones and regional lymph nodes, in contrast to our case where despite having a huge soft tissue tumor of the arm, evidence of extension to the bones was absent. None of the cases in this preliminary report had multifocal or distant metastatic disease. KHE may have variable locations. Although, most are commonly found as visible cutaneous lesions, lesions hidden within the retroperitoneum, abdomen, mediastinum, or cervicofacial regions5) and also regional lymph node metastases have been documented6). Distant metastases have not been reported in the course of KHE and there is a matter of debate whether it should be considered as multifocality or a true metastasis. Fernandez et al.7) have reviewed the English literature and finally reported 165 cases; among these 7 were in the chest wall, 4 in the mediastinum, one in the thyroid and one in thymus. Pulmonary involvement has not been reported in either of the reported cases. A thorough literature review has disclosed four reports of multifocal KHE with unusual presentations; a 3-year-old boy with submandibular lymphadenopathies, swelling of commisure of the lip and a lesion of right thyroid lobe8), a newborn with congenital cutanous multifocal KHE9), 3 children with multiple bone involvement10) and an infant with multiple bone lesions and bilateral cervical lymph nodes11). Chen et al.12) presented MRI findings of a histologically proven case of KHE of ankle in a 12-year-old boy and revealed multiple foci of the cutaneous tumor with different MRI morphologies along with intact underlying bones. The lesion also showed heterogeneous enhancement. Our case was similar to Chen et al.12)'s with respect to the absence of overlying skin changes, lack of invasion to the underlying bone and heterogeneous enhancement of the soft tissue tumor. An interesting aspect of our case was involvement of the pulmonary parenchyma which started as few small nodules with homogeneous enhancement. During a period of 6 months, the nodules increased both in size and number and displayed irregular necrotic centers with peripheral avid enhancement despite initial response to corticosteroids and interferon. Moreover, the patient developed bilateral pleural effusion with negative cytology (Fig. 2C). Given the patient's clinical and radiologic deterioration we decided to treat the patient with bevacizumab; a recombinant humanized antiangiogenic factor along with thalidomide. A search in PubMed identified a total of 6 case reports of epithelioid hemangioendothelioma with 5 cases of thoracic primaries that were treated with bevacizumab; one had partial response for 13 months, 1 had stable disease, and 4 had progressive disease13). An open-label, multicenter study demonstrated bevacizumab had been an effective and well-tolerated treatment for metastatic or locally advanced angiosarcoma and epithelioid hemangio endotheliomas14). Although, the patient initially responded to bevacizumab (Fig. 2D), after a short course of about two months, massive regrowth of pulmonary masses infiltrating normal lung tissues did occur (Fig. 2E, F). Another intriguing aspect of our case was absence of any evidence of consumptive coagulopathy or Kasabach-Merritt syndrome, a well-known association in KHE7).
In conclusion, according to the published literature, this was the first case of biopsy proven KHE of both lungs in a 13-year-old boy complicating soft tissue tumor of the extremity with further involvement of the spleen and multiple bones.
Acknowledgments
The authors would like to thank radiology department personnel; Mrs. Saeedi, Mrs. Ramezani and Ms. Taba for their kind efforts. We also appreciate outstanding care for the patient provided by nursing staff of the Pediatric Oncology Department of Mofid Children's Hospital.
Notes
No potential conflict of interest relevant to this article was reported.