Parry-Romberg syndrome with ipsilateral hemipons involvement presenting as monoplegic ataxia
Article information
Abstract
Parry-Romberg syndrome (PRS) is a rare, acquired disorder characterized by progressive unilateral facial atrophy of the skin, soft tissue, muscles, and underlying bony structures that may be preceded by cutaneous induration. It is sometimes accompanied by ipsilateral brain lesions and neurological symptoms. Here we present the case of a 10-year-old girl with right-sided PRS and recurrent monoplegic ataxia of the left leg. At 4 years of age, she presented with localized scleroderma over the right parietal region of her scalp; her face gradually became asymmetric as her right cheek atrophied. Brain magnetic resonance imaging revealed hemiatrophy of the face and skull base, and T2-weighted images showed increased signal in the right hemipons and hemicerebellar peduncle. Magnetic resonance angiography findings were unremarkable. She was treated with oral prednisolone, and her recurrent gait ataxia diminished within 2 months of the follow-up period. To the best of our knowledge, this is only the second case of PRS presenting with an abnormal involvement of the ipsilateral hemipons.
Introduction
Parry-Romberg syndrome (PRS), or progressive facial hemiatrophy, is a rare disorder presenting as a slowly progressive but self-limited atrophy of facial structures, sometimes followed by wasting of adjacent skin, connective or ocular tissue, muscle, cartilage, and bone1). Symptoms usually appear in the first or second decades of life23), but a few cases with a late onset have been described4). It slowly progresses over 2 to 20 years before stabilizing2). PRS is more common in females, and is believed to be sporadic, although some rare familial cases have been reported4).
Although some PRS patients are neurologically normal, diverse neurologic symptoms have been reported, most commonly seizures and headache5). Neuroimaging can disclose intracranial abnormalities, even in asymptomatic patients6). Common magnetic resonance imaging (MRI) findings include ipsilateral high signal intensity in the white matter or gray matter78), leptomeningeal enhancement7), calcification7), cerebral atrophy378), and intracranial vascular malformation9). Neurologic symptoms were not always associated with intracranial abnormalities5).
We present a unique case, a 10-year-old girl in whom PRS presented on the right face, who later had recurrent episodes of monoplegic ataxia. Brain MRI revealed very few findings with an increased signal at the ipsilateral hemipons and hemicerebellar peduncle.
Case report
The patient, a 10-year-old Korean girl, is the younger daughter of two children. She was born healthy and her family history was unremarkable. At the age of 4 years, her parents discovered mild hair loss over the right parietal region of her scalp, over an area of about 1 cm in diameter that slowly increased in size, centered around a small white papule (Fig. 1A, B). Diagnosis of localized scleroderma was made based on clinical features, and topical steroids were administered. The lesions slowly evolved over one year and became perpendicularly elongated with consequent atrophy, but without hyperpigmentation, and remained static after a year. Beginning from the age of 6 years, her face gradually became asymmetric atrophy on her right cheek, and worsened over the following two years. Unfortunately, the comprehensive evaluation was not performed by local orthopedic care despite her asymmetric facial atrophy. At the age of 10 years, she visited for sudden-onset ataxic gait on the left side two months prior. She had recurrent episodes of numbness, paresthesia, and limpness in her left leg, followed by falling down to the left side. Her arms were unaffected. These symptoms lasted one to two hours and spontaneously recovered. There was no history of headaches, visual dysfunction, or seizures and no history of recent viral infection or trauma. There was no family history of scleroderma, neurologic, rheumatologic, or autoimmune disease.
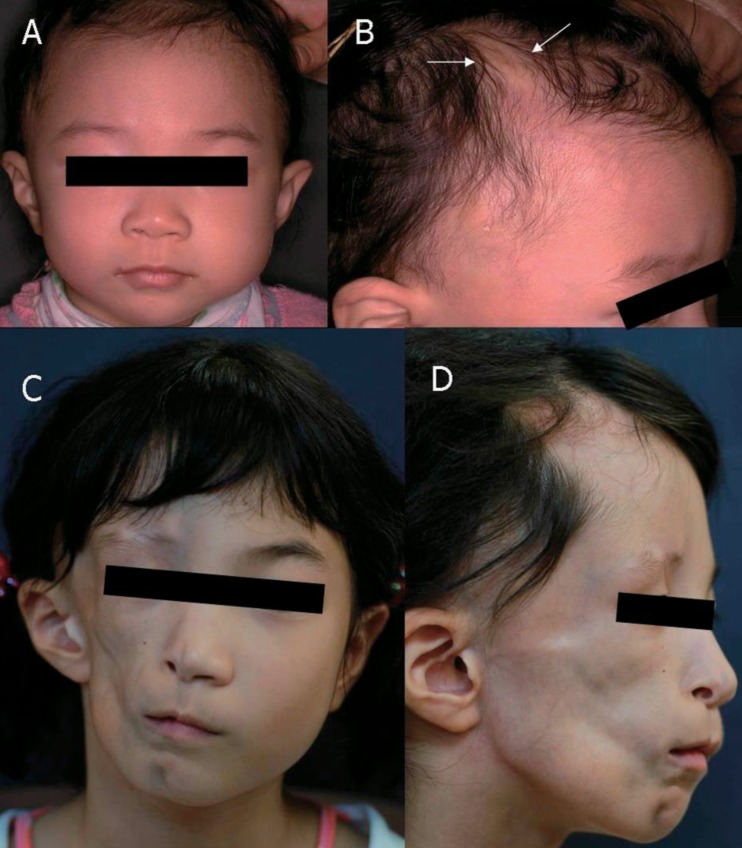
Photographic findings from our patient. (A, B) December 2002. At 4 years of age, the patient had hair loss over the right parietal region surrounding a small white papule (arrows), but had no abnormal facial features. (C, D) September 2007. At age 9, the patient presented with right hemifacial atrophy and linear scleroderma en coup de saber near the right paramedian frontal area, but without ptosis.
Physical examination revealed subcutaneous atrophy of the whole right hemiface, and a 3×0.5-cm linear scleroderma "en coup de sabre" (LSCS) in the right paramedian frontal area without skin discoloration (Fig. 1C, D). She was mentally alert and had normal intelligence. Meningismus was absent. Function of all cranial nerves was normal. Motor examination revealed 2 to 3 degrees of left leg weakness, but deep tendon reflexes were present in both legs without ankle clonus or Babinski responses. She showed deficits of the cerebellar function tests such as Romberg test, tandem gait, finger-to-nose test, and rapid alternating movements. General examination revealed no atrophy of the trunk on the same side or the ipsilateral limb.
Hematological and biochemical laboratory findings revealed normal values. Erythrocyte sedimentation rate and C-reactive protein were normal. Results of a hypercoagulable screen were negative. An autoantibody profile showed the presence of antinuclear antibodies at low titers (1:40, homogeneous). Rheumatoid factor was present at 19.2 IU/mL (range, 0-20 IU/mL). Antidouble stranded DNA, anti-Scl-70, lupus anticoagulant, anticardiolipin antibody, anticentromere, and test for syphilis (venereal disease research laboratory) were all negative. The cerebrospinal fluid was acellular, with normal protein and glucose. Oligoclonal bands were not detected. X-rays of the skull and face revealed no evidence of fracture, and abdominal sonography did not yield abnormalities.
MRI of the brain showed a hemiatrophy of right side facial structure and skull base underlying skin lesion. An increased signal was found at ipsilateral hemipons and hemicerebellar peduncle (Fig. 2). Magnetic resonance angiograms of the brain were unremarkable. An electroencephalogram did not show pathological findings. The diagnosis that best accounts for our patient's symptoms, disease course, and laboratory and MRI findings is PRS with recurrent monoplegic ataxia due to ipsilateral hemipons involvement.

(A, B) Brain magnetic resonance imaging (MRI) reveals right-sided facial hemiatrophy as well as bone defects on the right side of the skull base. (C, D) T2-weigthed MRI reveals hyperintensity in the right hemipons and hemicerebellar peduncle (arrows).
She was treated with oral prednisolone of 30 mg daily (1.2 mg/kg/day administered 3 times) for 2 months including a tapering period. Recurrent episodes of her ataxic gaits diminished within two months of follow-up period. Additionally, there was no progression of facial hemiatrophy or other neurological deficits for more than 3 years of observing the patient.
Discussion
First described by Parry (1825), the prevalence is unknown, but Stone4) suggested 1/500,000 births are positive for PRS. The disease usually begins during the first or second decades of life23). The progression of atrophy usually lasts from two to 10 years and then the process seems to enter a stable phase. The final condition of facial deformity may depend on the duration of the disease10).
PRS has been linked with many systemic manifestations including dermatologic, neurologic, ophthalmologic, cardiac, rheumatologic, infectious, endocrine, and maxillofacial fields. Seizures are the most common neurologic manifestation411). Seizures were documented as being medically intractable in 33% of patients5). Other symptoms associated with PRS were as follows: headaches412), movement disorders secondary to brain lesions1), trigeminal neuralgia12), hemiplegic migraines13), behavioral changes11), sympathetic hyperactivity12), progressive intellectual deterioration due to cerebral hemiatrophy (Rasmussen encephalitis)3), oculomotor nerve palsy11), facial nerve palsy11) and even death due to brainstem involvement13).
The most common findings on MRI are usually ipsilateral T2 hyperintensity, mostly in white-matter, gray matter and corpus callosum78). Other findings are as follows378910); leptomeningeal enhancement7), calcification7), cerebral atrophy378), intracranial vascular malformation9), focal corpus callosum infarctions7), ipsilateral infarcts in amygdaloid body7), and ipsilateral meningeal and basal ganglia lesions8). Neuroimaging findings in patients with PRS were frequently ipsilateral, although, contralateral findings have been reported7). To the best of our knowledge, only two cases of PRS with the T2 hyperintensity in the ipsilateral hemipons, including our patient, have been reported13). A two-year-old boy with PRS had many severe neurologic dysfunctions including seizures, dysarthria, oculomotor nerve palsy, and dysphagia. MRI revealed progressive atrophy of right cerebral hemisphere, and right midbrain and pons lesions. At the age of five years, he died in his sleep despite treatment with intravenous immunoglobulin and cyclophosphamide13). Our patient with PRS presented with recurrent episodes of monoplegic ataxia, and increased signal at ipsilateral hemipons and hemicerebellar peduncle on brain MRI. We presumed that the abnormal involvement of ipsilateral pons was associated with her symptoms. A follow-up MRI was planned after the treatment of steroid, but was not performed, because her parents were very concerned about recurrent use of sedative drug. We could not absolutely demonstrate, just might speculate, the correlation between a decline of ataxic gait and any improvement of neuroimaging.
The pathogenesis of PRS is not well understood and numerous etiologies have been suggested101415), including trauma, infection, autoimmune processes, and cervical sympathetic dysfunction. Autoimmune reaction appears to be the favored hypothesis in some patients, based upon coexisting autoantibodies15) such as antinuclear antibodies, antidouble strand DNA, anticardiolipin antibodies, rheumatoid factors, and oligoclonal bands in the cerebrospinal fluid, and good response to immunosuppressive therapy1). Inflammatory process, particularly vasculitis, is suspected in neurologic involvement of PRS. Brain biopsies have been performed in some children, revealing perivascular lymphocytic infiltrate, and gliosis and sclerosis of the leptomeninges13), suggesting an inflammatory process. Hyperactivity of the brain stem center has also been suggested as a cause of PRS14). In our patient, MRI discovered T2 hyperintensities at the ipsilateral hemipons, and her recurrent episodes of ataxic gaits gradually improved after the oral steroid trial, although all laboratory findings for autoimmune profiles and hypercoagulable screens were negative. Therefore, we speculated that abnormal autoimmune reaction and hyperactivity of the brain stem might be involved in her disease.
Chiu et al.6) reported a high rate of neurologic symptoms (28% to 65%) and neuroimaging abnormalities (19% to 73%) in children with PRS by institutional and literature review. However, neurologic symptoms were not correlated with neuroimaging abnormalities, and there was poor correlation between the severity of the cutaneous lesions and MRI findings. They recommended that all children with PRS or LSCS have a brain MRI performed at initial diagnosis, and that systemic immunosuppressive medication should be strongly considered for children found to have intracranial abnormalities. Early detection of neurologic involvement is important because it affects treatment options. Our patient was treated with oral prednisolone and the episodes of ataxic gaits gradually improved within two months without relapse or progression or other neurological deficits for more than three years of observing the patient.
We present a very curious patient with PRS who had recurrent monoplegic ataxia due to ipsilateral hemipons involvement without fatal neurological deterioration. Because clinical predictors of intracranial abnormalities are poor, neuroimaging should be strongly considered before starting treatment as a baseline examination to identify intracranial lesions and to determine therapeutic plans, although the children may be asymptomatic.
Notes
Conflicts of interest: No potential conflict of interest relevant to this article was reported.