Genetic risk factors associated with respiratory distress syndrome
Article information
Abstract
Respiratory distress syndrome (RDS) among preterm infants is typically due to a quantitative deficiency of pulmonary surfactant. Aside from the degree of prematurity, diverse environmental and genetic factors can affect the development of RDS. The variance of the risk of RDS in various races/ethnicities or monozygotic/dizygotic twins has suggested genetic influences on this disorder. So far, several specific mutations in genes encoding surfactant-associated molecules have confirmed this. Specific genetic variants contributing to the regulation of pulmonary development, its structure and function, or the inflammatory response could be candidate risk factors for the development of RDS. This review summarizes the background that suggests the genetic predisposition of RDS, the identified mutations, and candidate genetic polymorphisms of pulmonary surfactant proteins associated with RDS.
Introduction
Respiratory distress syndrome (RDS) is primarily due to the absence or transient deficiency of pulmonary surfactant (PS)1). RDS is a major acute postnatal pulmonary disease that especially affects the preterm newborns, and is caused by an insufficiency of PS metabolism. The disorder can progress to respiratory failure. Although the incidence of RDS is inversely related to gestational age, prematurity alone does not determine the risk of developing the disorder2). Besides surfactant deficiency, diverse risk factors such as gender, ethnicity, and maternal diseases, which have interactions with one another, influence the development and the severity of RDS2,3,4).
According to empirical observation and epidemiologic studies concerning racial differences and familial tendency, genetic disposition has been suggested for the risk of RDS5,6,7). In spite of antenatal steroid administration, postnatal surfactant therapy, and optimal ventilator care, not all infants of the same gestational period respond equally to treatments. From the intractable RDS cases, a few mutations, including genes encoding surfactant proteins B (SP-B)8), SP-C9), and the adenosine triphosphate-binding cassette transporter A3(ABCA3)10), have been identified. Specific polymorphisms related to surfactant structure and function, pulmonary differentiation, and inflammatory response could be candidate genes for the development of RDS2,11,12,13).
The present review summarizes the genetic aspect of RDS as follows. First, the histological and epidemiologic backgrounds that suggest the genetic predisposition for the development of RDS are introduced; these include studies on ethnic differences, and familial and twin studies. Then, the rare but revealed genetic causes of RDS are described, including the identified surfactant-associated mutations. Lastly, recent studies associating genetic polymorphisms for SP with RDS are shown.
Historical studies that suggest a genetic predisposition to RDS
1. Ethnic/racial differences
A higher rate of neonatal death and RDS has been reported for white newborn infants compared with black newborns, in spite of the relatively lower birth weights of the black infants5). The study of the US national mortality statistics for RDS in 1968-1973 showed a higher mortality rate in male infants than in female infants, and a lower incidence of fatal RDS in black preterm infants3). On the other hand, there was a report that showed no racial differences in fetal lung maturation between the blacks and whites in the US; the influence of environmental factors, such as maternal body weight and smoking, for the development of RDS was suggested14). In spite of these few opposing results14), regression analyses have shown that RDS occurs less frequently and less severely in black preterm infants, whereas white ethnicity is an independent risk factor of RDS in preterm and term infants15,16,17).
To date, the underlying mechanisms related to the racial difference in the occurrence of RDS have not been definitively revealed. Olowe and Akinkugbe18) suggested the earlier pulmonary maturation in Africans as being one of the mechanisms for the racial difference, based on the result of a higher amniotic fluid lecithin/sphingomyelin (L/S) ratio in African babies than in North American babies. Antenatal steroid treatment proved to be effective in non-Caucasians, whereas Caucasians showed little benefit from this intervention19). Richardson and Torday20) stratified the data by L/S ratio to adjust for possible differences in maturational timing between races, and showed lower rates of RDS in the black infants at every level of the L/S ratio. They suggested an alternative hypothesis of qualitative differences in the surfactants between races, including differences of phospholipids and surfactant proteins, or anatomic differences in the alveolar size and structure. Genetic differences such as allelic variation may contribute to the protein structure and function2,12,21).
2. Family studies
The increased risk of developing RDS with a history of previously affected infants has been suggested an important genetic or other familial tendency in its origin6,7). There should be other considerations for the results analysis of the family studies; that is, the possibility of including populations with a genetic predisposition to preterm birth, or the confounding environmental effects, as well as genetic influences2,6). Although most of the identified mutations of SP-B and ABCA3 are family specific22,23), and differences of SPs or maternal hormonal factor were suggested6), the integrative precise mechanism to explain these results is not clear.
3. Twin studies
Genetic predisposition in the susceptibility to RDS has been suggested by twin studies. The concordance difference of RDS between monozygotic (MZ) and dizygotic (DZ) twins (viz., the higher frequency of RDS in MZ than in DZ twins) supports the genetic contribution to RDS13,24). On the other hand, there was a study showing no significant concordance difference between MZ and DZ twins25). Several reports pointed out that concordance twin studies need to consider the differences in the mean gestational age, birth weight, and gender between MZ and DZ twins, as well as birth order and intrauterine environmental factors2,13,25).
Identified mutations of surfactant metabolism
SP-B and SP-C contribute to the surface tension-lowering activity of the PS. SP-B and SP-C are encoded by a single gene, located on chromosomes 2 and 8, respectively26,27,28). ABCA3, which is a transporter of phospholipid into lamellar bodies (LBs), the storage organelles for the surfactant complex, is critical for the proper formation of LBs and for surfactant function. Mutations in the genes encoding SP-B (SFTPB), SP-C (SFTPC), and ABCA3 (ABCA3) disrupt surfactant function and cause respiratory distress in the newborn11,23,29). Table 1 shows representative mutations of SFTPB, SFTPC, and ABCA3.
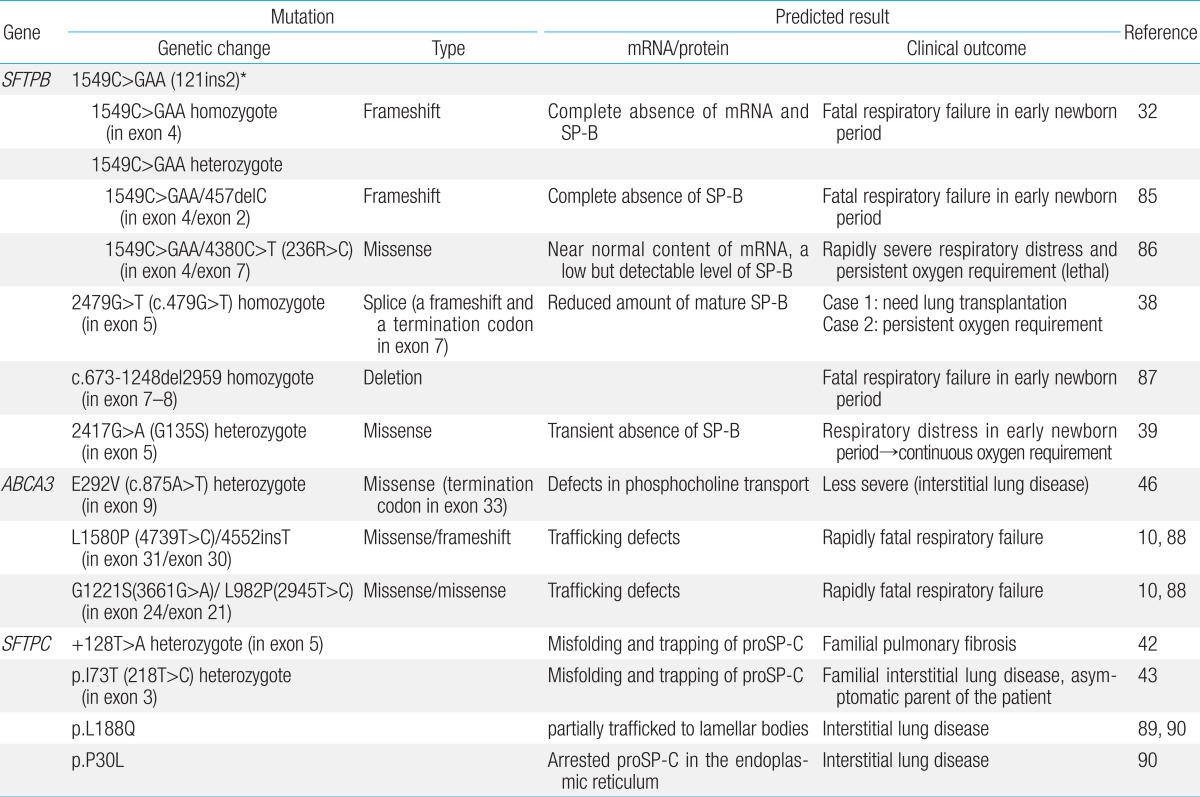
Representative mutations of SFTPB, ABCA3, and SFTPC
1. Hereditary SP-B deficiency
SP-B deficiency was first described in two siblings with severe lung disease in the newborn period8). Infants with hereditary SP-B deficiency, who are typically full-term, present severe respiratory failure shortly after birth, which is similar to RDS of preterm infants30). The most common mutation, found in 60%-70% of the cases of SP-B deficiency31), is a 1 base-pair (C) deletion and 3 base-pair (GAA) insertion, which is a net 2 base-pair insertion at codon 121 in exon 4 of SFTPB(termed 121ins2)32). It causes a frame shift and premature termination signal of translation, which results in absence of mature SP-B production. Infants homozygous for this autosomal recessive loss-of-function mutation are refractory to surfactant replacement therapy, and the cases are mostly fatal without lung transplantation33). The frequencies of 121ins2 are rare: 0.4% of a cohort in Missouri, 0.1% in Norway, and 0% in Korea and South Africa, in a population-based study34). Besides the 121ins2 mutation, more than 30 loss-of function mutations in SFTPB have been identified to date11,22,23,35,36,37). Other loss-of-function mutations appear to be family specific and sometimes associated with the partial or transient deficiency of SP-B that results in a more gradually progressive and prolonged survival38,39).
2. Mutations in SFTPC
The SFTPC gene encodes SP-C, a hydrophobic protein like SP-B, which functions to lower the surface tension of the PS40). The surfactant protein C precursor (proSP-C) is an essential endoplasmic reticulum (ER) membrane protein with an α-helical transmembrane domain. Various mutations in the ER-luminal C-terminal domain of proSP-C have been reported41). Mutations in SFTPC are thought to result in the production of misfolded proSP-C molecules that accumulate within the alveolar type II cells and cause cellular injury. Contrary to SP-B, mutations in SFTPC penetrate variably, and its expression ranges from severe RDS of the newborn to chronic interstitial lung disease of the adult or even asymptomatic conditions9,42,43,44). Presentation in full-term infants in the newborn period is associated with signs and symptoms typical of RDS, and may be fatal in the neonatal period45). Over half of the mutations are thought to arise spontaneously and result in sporadic cases23). About 35 dominantly expressed mutations in SFTPC have been identified, where threonine substitution for isoleucine in codon 73 (I73T) is the most common43).
3. Mutations in ABCA3
Several mutations of ABCA3 were first identified in 16 of 21 patients with severe neonatal surfactant deficiency10). Markedly abnormal LBs were observed by ultrastructural examination of lung tissue from four patients with different ABCA3 mutations. The clinical manifestation and course of ABCA3 mutations are variable: mutations in this gene can result in fatal surfactant deficiency in term newborn infants29), which have similar clinical and radiologic findings to SP-B deficiencies, and chronic interstitial lung disease in older children46,47). More than 70 mutations have been reported, most of which are family specific23). The overall frequency of mutations and disease is unknown.
4. Transcription factors that regulate the gene expression of surfactant proteins: mostly animal experimental studies
A transcription factor is a protein that binds to specific DNA sequences to control transcription to messenger RNA. A number of transcription factors, including thyroid transcription factor-1 (TTF-1; also known as NK2 homeobox 1 [Nkx2.1] or thyroid-specific-enhancer-binding protein [T/EBP])48,49), CCAAT enhancer binding protein-α(C/EBPα)50), and forkhead box A2 (Foxa2)51), have been identified that influence lung formation and surfactant production as well as homeostasis in late gestation. TTF-1, C/EBPα, and Foxa2 interact reciprocally to regulate transcriptional targets of surfactant synthesis and peripheral lung maturation50).
TTF-1, encoded by NKX2-1, is expressed in the thyroid gland, brain, and lung48). In the lung, it is an early marker of lung differentiation and an essential regulator for the expression of SP-A, SP-B, SP-C, and ABCA349). Mutations in NKX2-1 result in RDS and respiratory failure in newborn infants and in interstitial lung disease in older children36,49,52).
C/EBPα, a member of a family of basic leucine zipper transcription factors, plays an important role in the synthesis and metabolism of surfactant lipids and proteins. Deletion of the CEBPA gene causes the inhibition of lung epithelial cell proliferation and differentiation50).
Deletion of FOXA2 inhibits branching morphogenesis and epithelial cell differentiation of the lung51). Foxa2 modulates gene expression by interacting with other transcription factors. An animal study showed that the mammalian sterile 20-like kinase 1 and 2 (Mst1/2) regulated Foxa2, and might be crucial factors in surfactant homeostasis53).
Candidate genetic polymorphisms of SPs
1. SP-A
Among the three forms of PS54) (viz., intracellular and extracellular LBs, extracellular tubular myelin [TM], and surface monolayer), TM was absent in the lung of newborns who had died of RDS55). Moreover, the expression of SP-A was very low56). The levels of SP-A in tracheal aspirates, cord blood, and serum of newborn infants with RDS were lower than in infants without RDS57,58,59,60). Owing to these significant findings and the important roles of SP-A in the pulmonary host defense mechanism and the formation of TM61,62,63), the genes expressing SP-A have been thought to be strong candidates for susceptibility to RDS.
Two functional genes encoding SP-A (SFTPA1 and SFTPA2) are located on chromosome 10q22-q2364). Various single nucleotide polymorphisms (SNPs) have been discovered in coding regions and untranslated regions of SFTPA1 and SFTPA2 65,66,67), and many association studies with RDS have been reported68,69,70,71,72,73). Lee et al.74) and Kim et al.75) have presented the distribution and frequency of SP-A alleles in Korean newborns.
The SP-A1 6A2, SP-A2 1A0 variant and SP-A1/SP-A2 6A2/1A0 haplotype have frequently been reported as being associated risk factors for the development of RDS, and the SP-A1 6A3 variant as a protective factor for RDS, in Finnish and North American studies12,68,69,70). However, these association patterns were significantly different in a study of Korean preterm infants71). The SP-A2 1A0 variant and 1A0/1A0 genotype were associated with protection from RDS, whereas SP-A1 6A2 was not. The contrasting association of RDS with specific SP-A variants among different ethnic groups suggests that RDS is a multifactorial disease. In certain studies, associations between specific variants and RDS risk were confirmed under specific conditions or subgroups, such as infants born of twins or specific gestations, having the specific SP-B genotype or specific haplotypes of SP-A and SP-D69,72,73). Although various association studies of SP-A genetic polymorphisms and RDS have been reported, the precise mechanism of the cause-and-effect relationship between specific polymorphisms and RDS has not been revealed up to now12). Table 2 shows representative genetic polymorphisms of SPs associated with RDS.
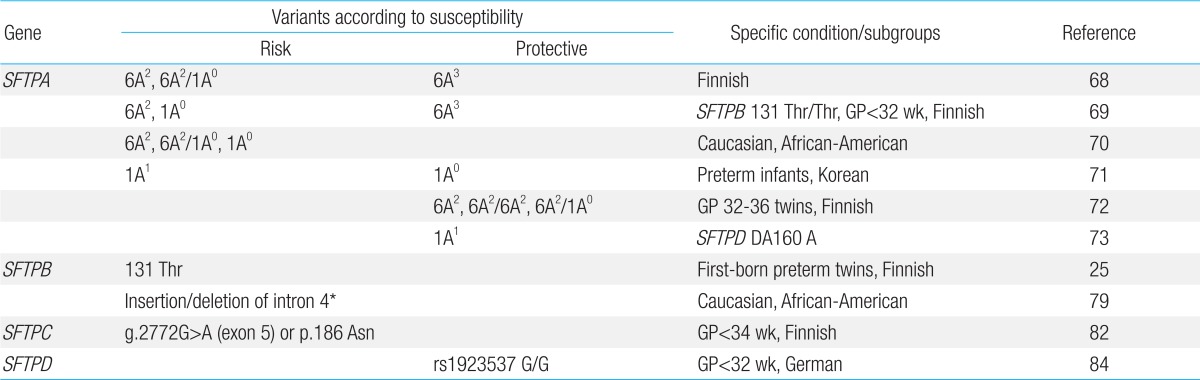
Representative genetic polymorphisms of surfactant proteins associated with respiratory distress syndrome
2. SP-B
SP-B plays essential roles in forming LBs and maintaining the surface monolayer76). SFTPB has been thought to be an important candidate gene for RDS. Hereditary SP-B deficiency caused by mutations of SFTPB results in lethal neonatal respiratory disease. The reduction of SP-B concentration to <25% of normal levels caused respiratory failure in adult mice77). SP-B and SP-C levels of tracheal aspirate were correlated with surfactant function in premature infants78). The SFTPB gene on chromosome 2p12-p11.2 is approximately 9.7 kb long, with 11 exons and 10 introns, including a large untranslated region26), and is polymorphic79,80). Hamvas et al.80) reported 86 SFTPB polymorphic sites (9.8/1,000 base pairs of SFTPB reference sequence), including 81 SNPs and 5 small insertion/deletions. Several studies showed specific SNPs or length variation (insertion/deletion polymorphic changes) in intron 4 of SFTPB to be associated with RDS development25,79,81).
3. SP-C
SP-C is expressed as a proSP-C that matures to SP-C, containing only 35 amino acids. SFTPC is located on the short arm of chromosome 8 (8p23.1)27). Mutations of SFTPB can disturb the proSP-C processing28). SP-C is thought to be a hydrophobic protein critical to lowering surface tension by enhancing the adsorption and spreading of surfactant phospholipids to the pulmonary surface monolayer; however, the exact functional roles of SP-C are currently not completely understood. Moreover, studies concerning the distribution of allelic variation in SFTPC and the associations with human disease are very rare. Lahti et al.82) showed that SP-C polymorphisms were associated with RDS risk among infants born at <28 weeks of gestation.
4. SP-D
SP-D is an important component of the innate pulmonary immune system and, like SP-A, plays a role in PS homeostasis61,62). The SFTPD gene, encoding for SP-D, is assigned to chromosome 10q22-q23 near SFTPA264). Specific polymorphisms of SFTPD influence the structure, function, and serum concentration of SP-D21,83). Associations of protective haplotypes of SP-A/SP-D with RDS development73) and specific genotypes of SP-D with the severity of RDS84) have been reported.
Conclusions
Because RDS is thought to be a multifactorial and/or multigenetic disease, it is necessary to be well-informed of the genetic contributors for RDS before designing any studies to search for new factors associated with the disorder.
To date, the frequency of inherited mutations that result in defective surfactant metabolism has not been revealed in the Korean population. Although it seems to be rare, the clinical outcomes of such mutations are mostly fatal. Therefore, we need to investigate the possibility of these mutations and to try to diagnose for them using genetic sequencing in neonatal cases of RDS presenting with an unusual course of severe respiratory failure, especially those with a familial history.
Even though the direct association of genetic polymorphisms with the development of RDS has not been confirmed to date, there have been many meaningful association studies. Large-scaled association studies are needed, considering the low penetrance of common genetic variants. The results of association studies can provide basic information for high-throughput genomic and proteomic researches and help to target specific therapies in the future.
Notes
No potential conflict of interest relevant to this article was reported.