Long-term outcome of patients with p22phox-deficient chronic granulomatous disease on Jeju Island, Korea
Article information
Abstract
Purpose
This study investigated the long-term clinical outcomes of patients with p22phox-deficient chronic granulomatous disease (CGD) on Jeju Island and retrospectively evaluated the effects of interferon-gamma (IFN-γ) prophylaxis.
Methods
The medical records of 15 patients with CGD were retrospectively reviewed. The efficacy of IFN-γ prophylaxis was evaluated by comparing the frequency of severe infections before and after starting continuous prophylaxis with IFN-γ.
Results
At the time of the analysis, 14 patients were alive, with a median age of 14.3 years. The diagnosis of CGD was made at a median age of 2.4 years, and the median age at onset of severe infection was 0.3 years. Thirteen of the 15 patients had their first severe infection within the first year of life. The overall incidence of severe infection was 1.36 infections per patient-year; pneumonia, suppurative lymphadenitis, and skin and subcutaneous abscesses were the most common infections. Aspergillus species were the most frequently isolated microorganisms, present in 15.8% of isolates. IFN-γ did not significantly change the rate of severe infection. The survival rate for patients after 2 years of age was 93%; there was a prolonged survival plateau beyond the age of 2.
Conclusion
Compared with cases of X-linked CGD reported in other studies, patients with CGD on Jeju Island did not show obviously different clinical manifestations, but they had a significantly higher survival rate. Further studies with a substantially longer period of observation, and with more patients under intensive surveillance are necessary to elucidate the prophylactic efficiency of IFN-γ.
Introduction
Chronic granulomatous disease (CGD) is an inherited primary immunodeficiency resulting from defects in any one of the subunits of the phagocyte nicotinamide adenine dinucleotide phosphate (NADPH) oxidase1). The NADPH oxidase complex is composed of five major subunits. Two of these, gp91phox (phox for phagocytic oxidase) and p22phox, are membrane-bound components encoded by the CYBB and CYBA genes, respectively. The remaining three components include p47phox, p67phox, and p40phox, encoded by the corresponding genes, neutrophil cytosolic factor 1 (NCF1), NCF2, and NCF42). The NADPH oxidase complex catalyzes the reaction of molecular oxygen to superoxide and related reactive oxygen intermediates (ROIs). Therefore, the functional activity of NADPH oxidase is significantly diminished or completely absent in patients with CGD, resulting in very low or no production of superoxide derivatives, which are important in killing invading microorganisms3,4). The X-linked form of CGD is caused by CYBB mutations, and accounts for about 70% of cases. The autosomal recessive (AR) forms are caused by mutations in CYBA, NCF1, and NCF2, accounting for about 5%, 20%, and 5% of cases, respectively5,6,7). Only one case with a mutation in NCF4 has been reported to date8).
The incidence of CGD is about 3-4 per 1,000,000 individuals1,3,4,9), and the prevalence of CGD in Korea is similar to other regions (3.4 per 1,000,000 individuals)10). On the other hand, the prevalence of CGD on Jeju Island is 20.7 per 1,000,000 individuals, which is 10-50 folds higher than in other regions of Korea8). We hypothesized that the high prevalence of CGD on Jeju Island was associated with the same mutation inherited from a common proband; all the patients with CGD tested on Jeju Island had an identical and homozygous mutation in the CYBA gene; c.7C>T in CYBA exon 1, and p.Q3X in p22phox 9). All patients with CGD on Jeju Island were presumed to be the A220 phenotype (where A indicates AR, and the superscript 0 indicates a complete absence of the affected subunit)11,12).
To date, few clinical studies on p22phox-deficient CGD have been carried out. In this study, we investigated the long-term clinical outcomes of patients with p22phox-deficient CGD on Jeju Island and retrospectively evaluated the effects of prophylaxis with interferon-gamma (IFN-γ).
Materials and methods
1. Patients
The medical records of Jeju National University Hospital from 2001-2012 were reviewed, and 18 patients with a diagnosis of CGD were identified. Diagnosis of CGD was initially based on abnormal granulocyte function tests, evaluated by a nitroblue tetrazolium test or a dihydrorhodamine-1,2,3 flow cytometry, and confirmed by Western blot analyses and mutation analysis. Because 3 patients had no available continuous follow-up data, 15 patients (6 males, 9 females) who had been seen regularly at the hospital were included in this study. This study was approved by the Institutional Review Board of Jeju National University Hospital (2013-10-002).
2. Clinical manifestations
Clinical presentations were reviewed retrospectively from the medical records of 15 patients with CGD. All severe infections were described according to the site of infection and the infectious agents that were isolated from blood, urine, stool, sputum or nasal aspirate, or aspirates of wounds or abscesses. A severe infection was defined as an episode of infection requiring hospitalization and intravenous antimicrobials or surgical treatment. To provide more accurate comparisons when follow-up time differed among groups, severe infections were presented as incidence per patient-year. Patient-years were calculated by adding all of the years that patients were followed in this study and multiplying by the total number of patients.
Chronic conditions, such as hepatosplenomegaly, being underweight or of short stature, anemia of chronic disease, and elevated liver enzymes, were also described. Hepatosplenomegaly was detected by physical examination or surveillance abdominal ultrasound. Being underweight or of short stature was defined as a condition in which the weight or height, respectively, of an individual was more than 3 percentile below the corresponding mean weight or height for a given age and sex. Anemia of chronic disease was regarded as anemia unresponsive to iron therapy, reduced levels of serum iron, serum transferrin, and total iron-binding capacity, or an increased level of serum ferritin.
3. The efficacy of IFN-γ prophylaxis
Patients with a body surface area of >0.5 m2 received IFN-γ at a dose of 50 µg, whereas those with a body surface area of <0.5 m2 received a dose of 1.5 µg/kg. IFN-γ was administered subcutaneously three times weekly. The efficacy of IFN-γ prophylaxis was evaluated by comparing the frequency of severe infections before and after starting continuous prophylaxis with IFN-γ.
4. Statistical analysis
The occurrence of the first severe infection and survival rates were analyzed by the Kaplan-Meier method. The Mann-Whitney U test was used to compare differences between the group receiving prophylaxis with IFN-γ and the group without IFN-γ prophylaxis, whereas the Wilcoxon signed-rank test were used to compare the frequency of severe infections before and after prophylaxis with IFN-γ. Statistical processing was carried out with IBM SPSS Statistics 20.0 (IBM Co., Armonk, NY, USA). P values <0.05 were considered to indicate statistical significance.
Results
1. Patients
Fourteen patients were alive at the time of analysis, with a median age of 14.3 years (range, 1.1-25.2 years). All 15 patients had an identical and homozygous mutation in the CYBA gene: c.7C>T in CYBA exon 1, p.Q3X in p22phox. The mean follow-up duration per patient was 8.4 years (range, 0.2-12.0 years).
Seven patients (47%) belonged to five families with multiple affected siblings, including four pairs of male-and-female siblings and one pair of female siblings. Three patients among these five families had died before the observation period.
2. Clinical manifestations
The diagnosis of CGD was made at a median age of 2.4 years (range, 0.1-13.1 years); in 7 of the 15 patients (47%) the diagnosis was made before the age of 1 year, and 5 patients (33%) were diagnosed before the age of 5. Only three patients were diagnosed over the age of 5 years (5.1, 7.2, and 13.0 years, respectively). The median age at the onset of severe infection was 0.3 years (range, 0-14.2 years). Thirteen patients (87%) with CGD experienced their first severe infection within the first year of life (Fig. 1).
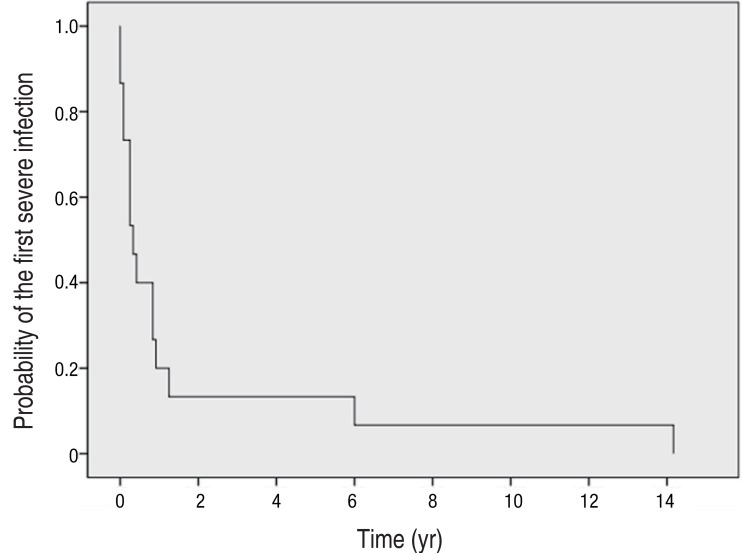
Probability of first severe infection. A severe infection was defined as an episode of infection requiring hospitalization and intravenous antimicrobials or surgical treatment. Thirteen patients (87%) with chronic granulomatous disease experienced their first severe infection within the first year of life.
At diagnosis, the majority of patients 47% (7/15) presented with lymphadenitis. Pneumonia was observed in 33% (5/15) of patients, and perianal abscess, gastrointestinal infection, and fever of unknown origin were noted for 7% (1/15) of patients. The most frequent types of first severe infection were similar to those at the time of diagnosis: lymphadenitis in 47% (7/15), perianal abscess (20%), pneumonia (20%), and fever of unknown origin (13%).
3. Localization of severe infections
The total number of severe infections recorded was 171, and the total period of observation was 126.1 patient-years. The overall incidence of severe infection was 1.36 infections per patient-year. Pneumonia was the most common severe infection, occurring in 32% of patients. Suppurative lymphadenitis was the second most frequent infection, occurring in 29% of the patients. Skin and subcutaneous abscesses were also reported in a considerable number of patients (16%). Other infections reported during follow-up are shown in Table 1.

Localization, frequency and isolated infectious agents from severe infections
4. Infectious agents of severe infections
Infectious agents were isolated in 19 of the 171 infections (11.1%) listed in Table 1. In most cases, no infectious agents were detected using proper procedures. Aspergillus species was the most frequently isolated microorganism, found in 15.8% of the isolates (3/19), resulting in pneumonia. Candida species were responsible for pneumonia and septicemia with a positive blood culture (each in one case). Staphylococcus aureus was isolated in suppurative lymphadenitis and septicemia with a positive blood culture (each one case). Other microorganisms were gramnegative, such as Pseudomonas species, Klebsiella pneumoniae, Salmonella species, Serratia marcescens, Enterobacter cloacae, and Burkholderia cepacia. Unusual pathogens, such as atypical Mycobacteria, were isolated from one case of suppurative BCGitis. Among the pneumonia cases, Mycoplasma pneumoniae were identified in two cases, detected by mycoplasma antibody titer.
5. Chronic conditions
Patients with CGD may have various chronic conditions, resulting from chronic inflammation or recurrent infections. The most common condition was hepato-splenomegaly, which was present in all patients. Being underweight and short stature occurred in 93% and 53% of the patients, respectively. Anemia of chronic disease and elevated liver enzymes were seen in 80% and 53% of the patients, respectively, probably resulting from chronic inflammation or recurrent infections.
6. Survival rate
One of the 15 patients with CGD died during the observation period, at the age of 2 years. He died of cardiopulmonary failure as a consequence of recurrent pneumonia caused by Aspergillus species. The survival rate for all patients was 93% after 2 years of age, and there was a prolonged survival plateau beyond this time (Fig. 2). Three patients who were alive at the time of analysis were excluded from this study due to lack of sufficient follow-up data.
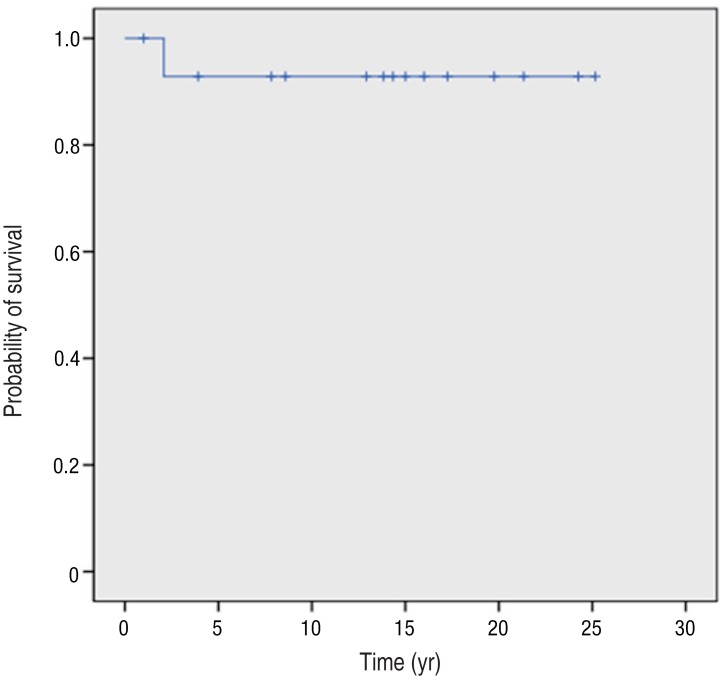
Kaplan-Meier survival curve of patients with chronic granulomatous disease on Jeju Island, Korea. The survival rate for all patients was 93% after 2 years of age, and the survival rate curve showed a prolonged survival plateau after 2 years of age.
7. Efficacy of IFN-γ prophylaxis
Seven patients received IFN-γ as prophylactic treatment, and eight patients did not receive IFN-γ prophylaxis for a variety of reasons, including the risk of systemic side-effects, noncompliance due to young age or inconvenience, and the burden of having to receive treatment for life; these latter patients were excluded from our study.
The overall observation periods were 36 and 77.9 patient-years for patients treated with and without IFN-γ, respectively. There were 35 severe infections in the group receiving prophylaxis with IFN-γ, compared with 117 in the group without prophylaxis, and no significant difference in the infection incidence per patient-year between the two groups (P=0.224) was observed (Table 2). The efficacy of IFN-γ prophylaxis was also evaluated by comparing the incidence of severe infections before and after starting continuous prophylaxis with IFN-γ. The observation periods were 12.2 and 36 patient-years for patients treated before, and after, IFN-γ prophylaxis, respectively. There was no significant difference in the infection incidence per patient-year between before and after IFN-γ prophylaxis (P=0.118) (Table 3). To detect the potential effects of IFN-γ in reducing certain types of infection, we analyzed the incidence of each severe infection in both groups of patients before and after IFN-γ prophylaxis. No significant differences in the reductions in certain types of infection were found (data not shown).
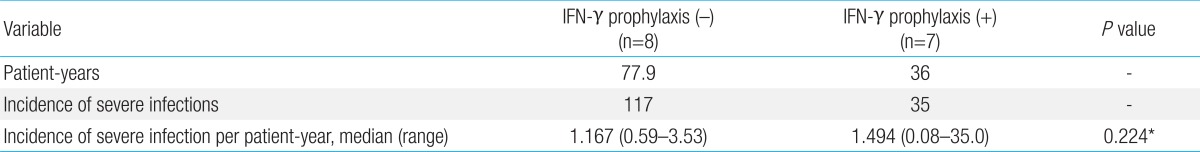
Effiectiveness of IFN-γ prophylaxis

Comparision of before and after starting prophylaxis with IFN-γ
Discussion
All patients with CGD on Jeju Island were presumed to be the A220 phenotype11,12). Therefore, we hypothesized that the prognosis of p22phox-deficient patients with CGD on Jeju Island would not be better than that of those with gp91phox-deficient CGD. Except for countries with high rates of consanguineous marriage, the proportion of p22phox-deficient CGD is smaller than other subtypes of CGD. Few reports have described the clinical presentation of p22phox-deficient CGD13,14). In a cohort in Israel13), which contained a relatively large number of patients with p22phox-deficient CGD (about 30%), similar to our cohort, most patients with gp91phox- and p22phox-deficient CGD were diagnosed earlier and had a shorter mean survival duration than patients with the other form of CGD. In our cohort, 87% of patients with CGD had their first manifestation within the first year of life. Similarly, 86% of gp91phox-deficient CGD patients in a Korean study had manifestations within the first year of life15). Furthermore, the incidence of serious infection per patient (11.4 in our cohort) was similar to that (10.3 in the Korean study) of gp91phox-deficient CGD15). Previously reported data regarding the age at diagnosis and first infection, localization of infection, and infectious agents in X-linked CGD followed a similar trend to our cohort1,9).
Since the first description of CGD nearly 60 years ago, the survival rate of patients with CGD has improved. This change may be attributed to better and earlier diagnosis of the disease, development of more effective antimicrobials, prophylaxis with antibiotics, antifungal drugs and IFN-γ, and bone marrow transplantation4). The overall survival rate of CGD is now thought to be about 90%4). Although a wide range of clinical variability in patients with CGD remains, X-linked CGD generally has an earlier and more severe clinical presentation, and a lower survival rate than the AR subtype. The prognosis of most patients with p22phox subtypes is typically poor, while it is better for those with p47phox-deficiencient CGD1,9). The prognosis may be dependent on the affected component of the NADPH oxidase complex, and the effect of the specific mutation on its residual activity.
However, patients with CGD on Jeju Island had a significantly higher survival rate compared with patients with gp91phox-deficient CGD in other studies (93% vs. <50% at 30 years of age)9,13,16,17). In the Israeli cohort, patients with X-linked CGD had a shorter mean survival time than p22phox-deficient CGD patients, as seen in our cohort13). Although these data should be interpreted with caution because of the small CGD subtype sample size, it appears that the specific NADPH oxidase gene mutation may not be linked to survival rates in patients with CGD. A recent study suggested that residual ROI production is more predictive of survival than the specific gene mutation in NADPH oxidase18). Even residual ROI production resulted in a significant gain in survival, and ROI production was not correlated with the expression of NADPH oxidase18). Interestingly, mutations in the flavin adenine dinucleotide- and NADPH-binding domains of gp91phox may allow normal protein expression, but little residual ROI production, and the amount of ROI production in patients with p22phox-deficient CGD, even with the A220 phenotype, varied widely18). Although residual ROI production may be an important parameter affecting the clinical course and survival, the variability in ROI production and the variable survival rate in patients with the same mutation cannot be explained at present. Further studies are needed to clarify whether residual ROI production can predict survival rates and to ascertain why ROI production does not correlate with NADPH oxidase expression.
The overall incidence of severe infections was 1.36 per patient-year in our cohort. This result is comparable with another retrospective study in Israel13), in which the annual infection incidence of p22phox-deficient CGD was 2.4 infections per patient-year before diagnosis and decreased to 0.7 infections per patient-year after diagnosis. All patients in the Israeli cohort received permanent prophylactic treatment with trimethoprim-sulfamethoxazole (TMP-SMX) and cephalothin or amoxicillinclavulanic acid. The decreased rate of infections was likely due to the fact that antibiotic prophylaxis significantly reduced the incidence of bacterial infection in CGD. In addition, until recently, fungal infections were the leading cause of mortality in CGD4), and several studies supported the efficacy of antifungal drugs against potential fungal infections in CGD4,9,13,17). In our cohort, most patients received prophylactic treatment with TMP-SMX and itraconazole from the point of clinical suspicion or observation of fungal infection, although prophylactic treatment was not taken by a large percentage of our patients at the time of analysis. Noncompliance might be due to alarm over potential side-effects of prophylactic antibiotics or the burden of taking drugs daily for life-long. Adequate support, such as regular check-ups by a physician, is needed to improve compliance with prophylactic treatment.
IFN-γ was shown to reduce the number and severity of infections in CGD patients in a large, multinational, multicenter, placebo-controlled study19). IFN-γ was effective for all genetic subtypes of CGD20). However, in our retrospective study, IFN-γ did not significantly change the rate of severe infections per patient-year or the incidence of infection between before and after IFN-γ treatment. Similar to our study, there was no additional benefit of IFN-γ as prophylactic treatment with cotrimoxazole and itraconazole in a prospective study in Italy17). The effectiveness of prophylaxis with IFN-γ is controversial, and the mechanism of how IFN-γ improves the host defense in patients with CGD remains unclear. Further studies with a substantially longer period of observation and significantly more patients under intensive surveillance and monitoring of compliance are necessary to clarify the prophylactic efficacy of IFN-γ.
Our study had several limitations. Because of its retrospective nature, this study could not precisely define the time at which patients suffered from the various infections. Multiple infections are sustained in patients with CGD by different strains of the same few species of bacteria21). We were able to detect such recurrences of mild infections in a large number of CGD patients. To compare clinical data among different cohorts of patients with CGD, we adopted severe infection as a measure to evaluate clinical manifestations and the efficacy of IFN-γ prophylaxis. In addition, we could not rule out confounding factors when analyzing the efficacy of IFN-γ prophylaxis, because we excluded the effects of antibiotic and antifungal prophylaxis. As mentioned above, a large number of the patients did not take their prescribed prophylactic treatments at the time of our analysis, further confounding the statistical analysis of the efficacy of IFN-γ prophylaxis.
Although the total number of patients in our cohort was small, and the follow-up duration was relatively short, we demonstrated that patients with p22phox-deficient CGD on Jeju Island showed no obviously different clinical manifestations from X-linked CGD as described in other studies, although they did have a significantly higher survival rate compared with patients with X-linked CGD. Further studies are needed to determine why CGD patients on Jeju Island have a higher survival rate despite their p22phox-deficiency. We suggest that early diagnosis, aggressive management of infections, continued intensive surveillance, and monitoring of compliance of prophylactic treatment will help to reduce the morbidity and mortality rates of patients with CGD.
Acknowledgments
This work was supported by a research grant from the Jeju National University Hospital Research Fund of Jeju National University in 2013.
Notes
Conflicts of interest: No potential conflict of interest relevant to this article was reported.