The strong association of left-side heart anomalies with Kabuki syndrome
Article information
Abstract
Purpose
Kabuki syndrome is a multiple congenital malformation syndrome, with characteristic facial features, mental retardation, and skeletal and congenital heart anomalies. However, the cardiac anomalies are not well described in the Korean population. We analyzed the cardiac anomalies and clinical features of Kabuki syndrome in a single tertiary center.
Methods
A retrospective analysis was conducted for a total of 13 patients with Kabuki syndrome.
Results
The median age at diagnosis of was 5.9 years (range, 9 days to 11 years and 8 months). All patients showed the characteristic facial dysmorphisms and congenital anomalies in multiple organs, and the diagnosis was delayed by 5.9 years (range, 9 days to 11 years and 5 months) after the first visit. Noncardiac anomalies were found in 84% of patients, and congenital heart diseases were found in 9 patients (69%). All 9 patients exhibited left-side heart anomalies, including hypoplastic left heart syndrome in 3, coarctation of the aorta in 4, aortic valve stenosis in 1, and mitral valve stenosis in 1. None had right-side heart disease or isolated septal defects. Genetic testing in 10 patients revealed 9 novel MLL2 mutations. All 11 patients who were available for follow-up exhibited developmental delays during the median 4 years (range, 9 days to 11 years 11 months) of follow-up. The leading cause of death was hypoplastic left heart syndrome.
Conclusion
Pediatric cardiologist should recognize Kabuki syndrome and the high prevalence of left heart anomalies with Kabuki syndrome. Genetic testing can be helpful for early diagnosis and counseling.
Introduction
Kabuki syndrome (KS) is multiple congenital anomaly malformation syndrome, first described by Niikawa et al.1) in 1981. The original description involved any patient with mental retardation, skeletal and auditory anomalies, cardiac defect, and distinctive facial appearance to the traditional make-up used by Japanese actors in the Kabuki theatre12). The cardinal features of KS include characteristic face, skeletal abnormalities, and dermatographic abnormalities, mental retardation, and postnatal growth deficiency3).
KS is rare disease. The prevalence of KS in the Japanese population is estimated as 1 in 32,0003). To date, this syndrome has been underdiagnosed outside of Japan. With increasing recognition of KS, reports of KS in various ethnicities have emerged4).
The molecular cause of KS had been unknown for a long time. Recently pathogenic variant in KMT2D and KDM6A were identified as the causative gene in KS. However the underlying genetic cause remained undiscovered in approximately 30% of KS patients.
KS is usually diagnosed by clinical findings, but early diagnosis is difficult, because typical characteristic face of KS is vague in neonate. Diagnosis can be delayed in children with congenital heart disease (CHD) who have typical Kabuki features due to lack of physician's knowledge about KS. In many cases, patients were misdiagnosed any other syndrome like Noonan syndrome, Turner syndrome, and Van der Woude syndrome. The relationship between KS and CHD is relatively well known. However, it is uncertain that which type of cardiac anomalies is frequently associated with KS.
This study was performed to analyze clinical features and cardiac defect and to investigate the predominant type of congenital cardiac anomalies in Korean KS in a single tertiary center.
Materials and methods
Between 2002 and 2014, 13 patients were clinically diagnosed as KS in our hospital. Each patient was evaluated by a clinical geneticist or cardiologists. The diagnosis of KS was based on the presence of characteristic facial configurations, skeletal anomalies, developmental delay, and anomalies of other organs. We retrospectively reviewed the medical records, laboratory studies, surgical notes, and genetic reports performed on each patient. Cardiac evaluation included echocardiography, electrocardiogram, chest x-ray, computed tomography, cardiac catheterization and angiography. Written informed consent was obtained from all participants or their parents before identifiable blood samplings for genetic studies. The study was approved by the Institutional Review Boards of Seoul National University Hospital (IRB No. 1404-115-575).
The data are expressed as median and range. Statistical analysis was performed with IBM SPSS Statistics ver. 19.0 (IBM Co., Armonk, NY, USA). Statistical significance of the relationship between growth retardation and KS was analyzed using Wilcoxon signed-rank test. And the statistical analysis of hypoplastic left heart syndrome (HLHS) as risk factor for death was tested using the Fisher exact test. P values of <0.05 were considered statistically significant.
Results
1. Demographic data
A total of 13 children with KS, aged 1 day to 12 years and 2 months were included. Their gestational age was 34+2 weeks to 40+1 weeks. Birth body weight was 1.86 to 3.99 kg. Six of them were male, and seven were female. Two patients died within 1 month after birth. Only four patients were diagnosed within 2 years of birth, with diagnosis further delayed in the remainder. The average age at diagnosis was median 5.9 years (range, 9 days to 11 years 8 months) and, the diagnosis was delayed by median 5.9 years (range, 9 days to 11 years 5 months) following the first clinic visit. They had been followed up for median 4 years (range, 9 days to 11 years 11 months) in our hospital. There was no patient with a family history of CHD or suspected KS.
2. Facial dysmorphism
The most recognizable feature of KS is the characteristic facial dysmorphism. All 13 patients had long palpebral fissures with everted lower eyelids. Arched eyebrow with sparseness of lateral one-third was another frequently observed finding. The most patients had short nasal septum and large ears, except two patients who died in neonatal period. Two patients had strabismus and one patient has ptosis of both upper eyelids. Two patients had high arched palate, and eight patients had a partial cleft palate including bifida uvula or cleft palate and/or lip, which required palatoplasty (Table 1).

Clinical manifestations among the 13 Kabuki syndrome patients
3. Noncardiac anomalies
KS showed multiple organ anomalies (Table 1). Ophthalmologic anomalies were presently evident in 6 patients (46%). These included ptosis, strabismus, and epiblepharon. Auditory problems such as hearing loss and middle ear effusion were observed in 10 patients (76%). Five patients (45%) showed renal anomalies including multicystic dysplastic kidney, duplex kidney, and single kidney. One boy had bilateral cryptorchidism requiring orchiopexy. Eleven patients (84%) had skeletal anomalies including brachydactyly, camptodactyly, and dislocation of the hip joints. Sacral dimple was observed in 7 patients (53%). Prominent fetal finger pad was identified in 9 patients (69%). Nine patients (69%) underwent noncardiac operations such as ventilator tube insertion on the tympanic membrane, palatoplasty for cleft palate, or reduction operation of developmental dislocation of hips. These operations were performed median 2 times (0-6 times) during the follow-up.
4. Congenital cardiac anomalies
CHDs were diagnosed in 9 KS patients (69%). All 9 patients showed left-side heart anomalies including HLHS in 3 patients, coarctation of aorta (CoA) in 4 patients, aortic valve stenosis and mitral valve stenosis in each 1 patient. No one had right side heart disease or simple isolated septal defect in our patients. (Table 2, Figs. 1, 2) The prevalence of left side heart diseases in our study was high in KS patients (9/9, 100%). Surgical interventions were necessary in 7 of 9 patients. Hybrid or Norwood procedures were performed in 2 patients with HLHS. One patient with HLHS completed the sequential procedures of Fontan operation at 4 years of age. The other one patient with HLHS died 22 days after stage I hybrid procedure. The remained one neonate with HLHS died after her parents refused surgical correction of HLHS. Coarctoplasty and defect-repair procedures were performed successfully in 4 patients.

Prevalence of cardiac defects with Kabuki syndrome in this series
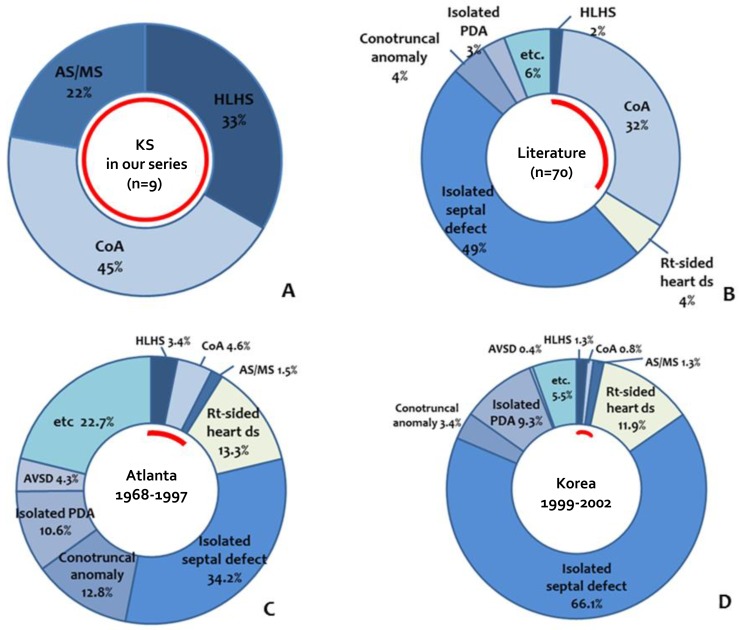
Prevalence of cardiac defects with KS in our series, the literature, and normal populations. (A) In our series, all patients with KS had left-sided heart anomalies. (B) In the literature, 49% of KS patients had isolated septal defects, and 34% had left-sided heart anomalies. (C, D) In the normal populations from Atlanta (USA) and Jeju (Korea), left-sided heart anomalies were found in 9.5% and 3.4% of the population, respectively. The red line indicates the percentage of patients with left-sided heart anomalies in each group. KS, Kabuki syndrome; AS/MS, aortic stenosis/mitral stenosis; HLHS, hypoplastic left heart syndrome; CoA, coarctation of the aorta; PDA, patent ductus arteriosus.

Congenital cardiac defects in Kabuki syndrome: left-sided heart anomalies. (A) Cardiac computed tomography reveals coarctation of the aorta (arrow) in patient 13 (B) The apical four-chamber view during echocardiography reveals mitral valve atresia and a hypoplastic left ventricle in patient 5. RV, right ventricle.
5. Molecular genetic characteristics in our patients
In our patients, total 10 patients except early expired 2 patients and unconsent 1 patient (76%) were conducted molecular genetic analysis. Whole exome sequencing was used as the method for mutation detection. KMT2D gene mutations were identified in 9 patients. There was no mutation in both KMT2D and KDM6A gene in 1 patient. All identified mutations were novel and there was no repetitive mutation. The most of them were nonsense mutations (6/9, 69.2%). Two small deletions and one small insertion cause translational frame-shifting and premature termination of protein synthesis7) (Table 3).

Summary of the 13 Kabuki syndrome patients
6. Intermediate follow-up result
Total 11 patients were followed up in our center for median 4 years (range, 9 days to 11 years 11 months). Patients were hospitalized median 5 times (range, 0-8 times) during follow-up. Three patients (23%) manifested as failure to thrive during follow-up period (birth weight <5 percentile). However our patients did not showed significant growth retardation as compared with birth body weight (P=0.091 by Wilcoxon signed rank test). One patient was required gastrostomy tube insertion for a feeding problem. In addition 10 of 11 patients (90.9%) showed evidence of developmental delay with mild to severe degree4). In our study, 3 patients showed intelligence-quotient below 70, and 3 patients received three-level developmental delay. Four patients were not evaluated by formal methods, but they appeared to have delays in speech and language development. Only 1 patient showed normal intelligence. Total 11 patients survived and 7 patients with congenital cardiac anomaly were alive now. HLHS was the most important risk factor for death (2/3 vs. 0/10; P=0.038 by Fisher exact test).
Discussion
KS is a multiple malformation/mental retardation syndrome4). It is well known that CHD are frequently accompanied by KS. The prevalence of CHD with KS varies widely from approximately 28% to 80%8). Shunt lesions or conotruncal abnormalities such as tetralogy of Fallot with KS have been in early series and left sided anomaly including HLHS with KS was more recently described891011). Although the various types of CHDs are well described for patients with KS, the dominant type of cardiac defects may be underestimated. It was the objective of this study to investigate the dominant type of CHD in KS.
In our series, CHD was present in 9 of 13 (69%), and all 9 had left-sided anomaly. Compared with the prevalence of heart anomalies in the previous studies conducted in Atlata6), the incidence in our series is much higher than in the normal population, with left-sided obstruction in 7% of patients, and HLHS in 2.1%. Similarly, the prevalence of left side heart disease in our study was higher than general Korean populations of prior studies in Jeju island in 3.1% (8/236)5).
The strong association of left-sided anomaly with KS was found in the current study. The previous study reported that CHD was present in 58% of 60 KS patients. CoA (22.8%) and septal defects (37%), such as atrial septal defect or ventricular septal defect were the most common type of CHD. Left-sided anomalies were evident in 28.5% of total KS patients with CHD8). Another report showed the prevalence of left-sided obstructions with KS was 31%, which was significantly higher than that of general population (14%)12). According to analysis of 121 patients with KS, CHD was present in 71 of 121 (59%) with KS. Of them, left-sided obstruction was present in 27 of 171 (38%) (Fig. 1)11).
Recently, the genetic studies of KS have been conducted towards causative gene mutations. In 2010, KMT2D (previously known as MLL2) at 12q13.12 was identified as the first gene associated with KS to be identified. Since then, 55% to 65% of KS cases have been reported to harbor the mutations in KMT2D13). KMT2D gene encodes a histone-3 lysine-4 specific methyltransferase (H3K4), that belongs to the SET1 family of histone methyltransferases (HMTs). H3K4 activates multiple genes that are important for embryogenesis, development, and cell differentiation14). H3K4 is part of a large protein complex called ASCOM, which is a transcriptional regulator of estrogen receptor genes15). Deregulated estrogen receptor mediated pathways are associated with a large spectrum of KS phenotypes, including cardiac anomalies16). H3K4 also acts with other proteins that, includes Wdr5, Rbbp5, Ash2l, and the histone-3 lysine-27 demethylase (H3K27) (encoded by KDM6A). Several studies have reported that KMT2D mutations are involved in genetic diseases, such as KS or cancers, such as medulloblastoma and, myeloma. Notably, a recent study has shown that KMT2D is involved in the control of proliferation and differentiation in mouse embryonic stem cells, particularly in cardiac-specific lineages17).
In 2012, a de novo deletion mutation and point mutations in KDM6A (previously known as UTx), which is located at Xp11.3, were identified as also being involved in KS18). From among KS patients without a KMT2D mutation, 9% to 13% had a mutation in KDM6A7). KDM6A encodes the H3K27, a JmjC domain containing protein, that plays a crucial role in cell proliferation and antagonizes polycomb group-mediated repression. KDM6A also functions in the ALR complex together with the product of KMT2D19). In recent studies, KDM6A knockdown fish exhibited craniofacial anomalies, including cleft palate20). In addition, KDM6A-deficient mice had severe heart defects21). KDM6A also affects the expression of key regulatory genes involved in adipogenesis and osteogenesis22). These observations support the hypothesis that regulatory pathways by KDM6A play an important role in KS.
Total 10 patients received genetic assay in our series. 9 of them were confirmed to have a KMT2D gene mutation. KMT2D and KDM6A mutations were absent in only a single patient. Five patients had been reported in the previous publication by Cheon et al.7). A recent report described that, 20% to 45% of KS patients remains unknown concerning the genetic basis of the syndrome18).
The increasingly recognized role of genetic factors in the etiology of CHDs has been shown by the high frequency of heart anomalies in genetic syndromes23). The correlation between cardiac anatomies and some genetic anomalies indicates that specific morphogenetic mechanisms set in motion by genetic mutations can result in specific cardiac phenotypes. Chromosomal anomaly or genetic syndrome with CHD can be considered an important staple resource for gene identification involved in cardiac morphogenesis23).
A typical example of the link between a genetic syndrome and CHDs is Turner syndrome. A large proportion of patients with Turner syndrome have CoA and bicuspid aortic valve. Turner syndrome is well known in subjects with monosomy X10). Sex chromosome anomalies have been observed in several KS patients, before the causative genes have been identified. In particular, features resembling KS were reported in a female patient with a ring X chromosome24). In addition, in previous studies of KS patients, left-sided heart anomalies, including CoA, were found predominantly in males8). Taken together, these observations indicate that, X-linked genetic factors may be involved in determining cardiac defects in some patients825). Notably CoA and left-sided anomalies were observed predominantly in males in the normal population. It is supporting the hypothesis that the presence of two X chromosome can prevent left-sided heart anomalies823). Additional evidence supporting this hypothesis is the fact that KDM6A, one of the causative genes for KS, is located on the X chromosome (Xp11.2)16). If the association of KS with left-sided heart anomalies is found to be significant and the genetic cause of KS is clearly revealed, we will obtain abundant knowledge about the mechanisms of cardiac development via the molecular study of this disease.
Nevertheless, a multifactorial mechanism is still remained in the etiology of left-sided anomalies. In our series, there was no male preponderance of left-sided obstruction lesion. But, this may have reflected that the small number of our series and the smaller proportion of KDM6A mutations in KS. In our study, we detected hypoplastic left heart anomaly is a high proportion of KS with CHD. There are needs to more molecular studies supporting mechanism of the high association between left-sided heart defect and KS. Authors agree that the data may be biased because this is a small series study of KS from a tertiary hospital. Our center is one of the major referral hospital for complex CHD, therefore the most severe form of cardiac malformation, such as HLHS, may be found more frequently.
The probability of long-term survival is partly determined by the presence of heart problems in KS patient. In this study, HLHS was the risk factor for death of KS. Most left-sided obstructive lesions have high mortality unless surgical correction in neonate. With the evolving surgical technique, the survival rate improved.
We believe that KS has been under-detected due to difficulties in early diagnosis and lack of molecular genetic analysis. Because cardiovascular presentation may be the earliest and the most obvious manifestation, the cardiologists are frequently the first physicians for KS patients. Therefore, pediatric cardiologist should have knowledge of KS and need to evaluate carefully about possibility of KS. Also they should be aware of high prevalence of left heart anomaly with KS, because cardiovascular presentation can be the first and the severe manifestation. On the other hand, the patients with clinical manifestation of KS are necessary for cardiac evaluation with echocardiography. It is suggested that genetic and developmental background related to cardiac malformation and KS may be clarified for prevention, further management and counseling.
Notes
Conflicts of interest: No potential conflict of interest relevant to this article was reported.


