Myelin oligodendrocyte glycoprotein antibody-associated disorders: clinical spectrum, diagnostic evaluation, and treatment options
Article information

Abstract
Inflammatory or immune-mediated demyelinating central nervous system (CNS) syndromes include a broad spectrum of clinical phenotype and different overlapping diseases. Antibodies against myelin oligodendrocyte glycoprotein (MOG-Ab) have been found in some cases of these demyelinating diseases, particularly in children. MOG-Ab is associated with a wider clinical phenotype not limited to neuromyelitis optica spectrum disorder, with most patients presenting with optic neuritis, acute disseminated encephalomyelitis (ADEM) or ADEM-like encephalitis with brain demyelinating lesions, and/or myelitis. Using specific cell-based assays, MOG-Ab is becoming a potential biomarker of inflammatory demyelinating disorders of the CNS. A humoral immune reaction against MOG was recently found in monophasic diseases and recurrent/multiphasic clinical progression, particularly in pediatric patients. This review summarizes the data regarding MOG-Ab as an impending biological marker for discriminating between these diverse demyelinating CNS diseases and discusses recent developments, clinical applications, and findings regarding the immunopathogenesis of MOG-Ab-associated disorders.
Key message
MOG antibody-associated disorder exhibits different pathophysiological and phenotypic findings than both aquaporin-4 antibody-associated neuromyelitis optica spectrum disorder and typical MS. MOG-antibody is of particular interest in pediatric patients with clinical or radiological non-MS typical findings. MOG-antibody was included in a diagnostic algorithm for children recommending for the first time a standardized use in clinical practice except in cases of typical MS.
Introduction
Inflammatory or immune-mediated demyelinating central nervous system (CNS) diseases are a heterogeneous group that includes mono- and multiphasic diseases with prognoses ranging from benign to fulminant and a variety of different treatment responses. The most common demyelinating CNS diseases have some chance of misdiagnosis that occurs in up to 10% of patients [1]. Diagnosis is based on a combination of clinical manifestation and radiological and laboratory findings. In 2004, the specific autoantibodies against aquaporin-4 (AQP4), a plentiful water channel on astrocytic endfeet in the CNS, in patients with neuromyelitis optica spectrum disorder (NMOSD) fortified the diagnosis of and research into demyelinating CNS diseases [2]. However, a subgroup of clinically definite NMOSD patients showed AQP4-seronegative results [3].
Myelin oligodendrocyte glycoprotein (MOG) is exclusively expressed in the CNS on the outer surface of the myelin sheath and oligodendrocyte plasma membrane [4]. MOG was comprehen sively studied as a potential target structure in CNS demyelinating diseases. The majority of MOG antibody (MOG-Ab)-seropositive patients have optic neuritis (ON), encephalitis with brain demyelinating lesions, and/or transverse myelitis (TM); the new term “MOG-Ab–associated ON, encephalitis, and myelitis” has been suggested to include these patients with CNS demyelinating syndromes and MOG-Ab positivity. Depending on the clinical assessment, patients can be clinically diagnosed with NMOSD, acute demyelinating encephalomyelitis (ADEM), multiple sclerosis (MS), or isolated ON or TM [5].
In the last few years, the MOG-Ab–associated disorder (MOGAD) spectrum has been rapidly broadening, and as more data regarding their clinical, radiological, and laboratory findings have become available, their immunopathogenesis has been elucidated. This review article discusses the evolving immunopathogenesis, clinical spectrum, diagnostic approach, prog nostic research data, and treatment principles for clinical practice.
MOG-immunoglobulin G: pathogenic role
MOG, a glycoprotein of the immunoglobulin superfamily, is a component of the CNS myelin sheath, as are myelin basic protein and proteolipid protein [6]. The precise functions of MOG remain to be clarified but likely include roles in the adhesion of myelin fibers, regulation of oligodendrocyte microtubule stability, and modulation of the interaction between myelin and the immune system by the complement pathway [7].
In humans, high-titer MOG-immunoglobulin G (MOG-IgG) levels in serum samples seem to efficiently activate the complement cascade in vitro [8]. Furthermore, purified IgG from MOG-IgG–positive patients, when incubated with oligodendrocytes in vitro, led to obvious cytoskeletal disorganization, further suggesting functional pathogenicity [9]. Initial studies revealed a pathogenic effect of the humoral immune response against MOG (human MOG-Ab); these antibodies were able to induce the death of MOG-expressing cells as well as natural killer cell-mediated cell death, with the extent of cell damage dependent on antibody levels [5]. In addition, MOG-Ab belongs to the complement binding IgG1 subtype and has been found to activate the complement cascade, leading to complement-dependent destruction of MOG-expressing cells [8]. The ac cumulation of MOG-Ab has been described in CNS antigen-presenting cells with subsequent activation of autoreactive T cells, followed by the induction of peripheral autoreactive T cells [10]. Significantly, MOG-Ab alone did not induce inflammation or tissue destruction; rather, their interdependence with T cells was required to develop their pathogenic potential [10].
Despite MOGAD’s ability to overlap with the clinical presentation of AQP4-IgG–associated NMOSD, the mechanisms of the two disease groups are likely different. While the pathological hallmark of AQP4-Ab–positive NMOSD is astrocytic damage with secondary oligodendrocyte loss and demyelination, no evidence of astrocytopathy has been reported in MOG-Ab positivity, which was related to the inflammation and myelin destruction affecting oligodendrocytes without astrocyte injury [11].
To our knowledge, several cases to date with obtainable neuropathology have been described in the literature [12-15]. Interestingly, most cases discovered MS pattern II lesions with obvious demyelination, marked infiltration of macrophages (containing myelin degradation products), and T cells, preoligodendrocytes, and relatively preserved axons and astrocytes. The inflammatory hallmark is an infiltrate consisting of T cells as well as a complement, B cell, and IgG [3] suggestive of a humoral pathogenesis. The clinical presentation of MOG-Ab–positive patients with MS pattern II pathology diverges and includes presentation with clinically isolated syndrome, MS, NMOSD, recurrent longitudinally extensive transverse myelitis (LETM), and atypical inflammatory demyelinating CNS syndromes [12-15].
Clinical presentation of MOGAD
Clinical MOG-Ab–positive patients can present with an NMOSD phenotype. Overall, in AQP4-negative patients, MOG-Ab had a prevalence of 25% in subsequent studies [3]. In contrast to AQP4-Ab–associated disorders with the well-defined phenotype of NMOSD, those with MOGAD have a less well-defined clinical presentation. The foremost findings that distinguish MOG-Ab from AQP4-Ab NMOSD are summarized in Table 1 [16].

Clinical and neuroimaging findings indicative of MOG-Ab versus AQP4-Ab in neuromyelitis optica spectrum disorder
1. Prevalence
The prevalence of MOG-Ab seropositivity among patients with NMOSD has been reported by several studies and varies widely depending on the inclusion criteria. Sato el al. [17] found that 7.4% of all NMOSD patients were seropositive for MOG-IgG, while 64.7% were seropositive for AQP4-IgG. When alterations are made for patients with specific phenotypes and a lack of AQP4-Ab, the proportion of MOG-Ab-positive patients becomes much higher. Two recent studies reported that 40% of patients with bilateral or recurrent ON and negative AQP4-Ab were positive for MOG-Ab [17,18]. Regarding AQP4-Ab-negative LETM, the described prevalence of MOG-Ab was 7.4%–23.2% [17,19]. Among children, that prevalence appears to be higher: 50% for patients with definite NMO and 80% for patients with recurrent ON [20].
Particularly, double positivity (i.e., for both AQP4-Ab and MOG-Ab) is usually not identified using cell-based assays, which suggests that each antibody is present in distinct disease processes. Isolated cases of double positivity are extremely rare and usually have significantly higher relapse rates, long-term dis ability, and magnetic resonance imaging (MRI) lesion bur dens, but these characteristics are more compatible with AQP4-Ab–positive NMOSD [21,22].
2. Demographic features
The proportion of males is generally higher (47%–62%) among MOGAD patients than among those with AQP4-Ab–positive NMOSD (only 10%–15%) [17,23]. However, a recent multicenter European study reported that 74% of MOG-Ab–positive patients were female, resulting in a female-to-male ratio of 2.8:1 [24]. However, this ratio of female predominance is lower than that of up to 9:1 reported in AQP4-Ab patients [25].
Regarding age at onset, some studies documented younger age (around the third decade) in the MOG-Ab group versus the AQP4-Ab group (the fourth decade), while others reported no difference [17,23]. A few studies presented the age-dependent clinical phenotype, with a predominance of ON found in adult-onset MOGAD compared to a ADEM-like patterns in children with better recovery [23,26,27]. Seizures and encephalitislike symptoms are more frequent in MOGAD than AQP4-Ab-positive disorders [28].
3. Clinical phenotypes
Initially, the MOG-Ab seemed to be associated with a monophasic course and predominantly present in children with an ADEM-like onset [5,29]. In subsequent studies, MOG-Ab were present in a subgroup of patients with ADEM, NMOSD, monophasic and relapsing ON, TM, and demyelinating syn dromes overlapping with anti-NMDA receptor encephalitis or glycine receptor alpha 1 subunit antibody-positive ON [5,23,29].
Two independent groups have evaluated the categories of clinical phenotypes for MOGAD: ON was the major phenotype (41%–63%), followed by LETM (29%–31%), NMO (6%–24%), and encephalomyelitis (2%–6%), while in AQP4-Ab-positive patients, the reported phenotypes were NMO in approximately 60%, followed by LETM (in about 30%), and ON (in about 10%) [17,24,30]. The first attack of TM or ON can be severe. In the largest series, visual acuity was described to be <0.1 at least once in 69% of patients with ON [24]. Among patients with TM, motor symptoms were frequent and included tetraparesis in 28%, paraparesis in 48%, and severe weakness in 21% [24]. Brainstem involvement and postrema area syndrome (persistent nausea, vomiting, or hiccups), which usually present in NMOSD (around 40%), has been reported in 6%–15% of MOG-Ab–positive patients as well [17,31]. In a large cohort of MOG-Ab–positive patients, ADEM (or an ADEM-like episode) was reported as the initial feature in 18%, primarily affecting children [31]. Cortical encephalitis was recently described in MOG-Ab-positive patients but not in AQP4-Ab NMOSD [28,32].
A humoral immune response against MOG is only rarely seen in MS [3]. MOG-Ab has been reported up to 5% of patients diagnosed with atypical MS: severe attacks of myelitis, ON, and/or severe brainstem syndromes, with failure to several disease-modifying drugs [33]. In this subgroup, frequent relapses and insufficient responses to immune therapy seem common.
MOGAD in pediatric patients
Several clinical conditions compatible with MOGAD have been described in pediatric patients with MOG-Ab positivity, mainly multiphasic ADEM, ADEM followed by ON, relapsing ON, TM, and AQP4-Ab–negative NMOSD [34]. Particularly in relapsing diseases, the differentiation of rare conditions—such as NMOSD, multiphasic ADEM or relapsing ON—has important therapeutic counsel. The first evidence for the role of anti-MOG-IgG as biological markers in children was identified by O’Connor et al. in a subgroup of ADEM [35]. Some studies have found that MOG-Ab-positive patients different clinical features according to age [36]. They found a bimodal distribution in 13 pediatric MOG-Ab-positive patients, with encephalopathy being more common in younger patients (4–8 years) and ON in older patients (13–18 years) [36].
Overall, 39% of MOG-Ab–positive children have a recurrent disease course versus only 5% with a typical MS disease course. The serial analysis of MOG-Ab–positive patients was recommended at 6–12 months. As there is no gold standard for MOG antibody analysis, particularly for children, the clinical and laboratory findings suggestive of MOGAD retesting is reasonable. As the MOG-Ab titers found in pediatric MS are usually lower and MS diagnostic criteria differ [37], there is no clear association between MOG-Ab and MS, even in pediatric patients.
Disease course
A preceding infectious episode was noticed in 47% (e.g., influenza, infectious mononucleosis, following vaccination), with median follow-up times of 12–24 months [38]. The proportion of patients with a single attack is likely reduced with long-term follow-up, in which the proportion of patients with a single attack was only 29% during a longer median follow-up time (43 months) [30]. The median time between the first and second attacks was longer in MOG-Ab–positive patients (11.3 years) than in AQP4-Ab-positive (3.2 years) or double seronegative (3.4 years) patients [29]. However, other study revealed the median time between the first and second attacks in MOGAD was 5 months, although the interval could be longer (more than 12 months in 8 patients and up to 492 months in one patient [24]).
Laboratory evaluation of MOGADs
1. MRI findings
On brain MRI, supratentorial lesions were found in the brainstem of nearly half of patients, while cerebellar lesions were noted in one third [24,27,39]. Lesions involving the deep gray matter and lesions adjacent to the fourth ventricle were noted more frequently in AQP4-Ab NMOSD than in the MOGAD [23]. It has become possible to distinguish MOGAD from MS with a specificity of 95% and a sensitivity of 91% by different lesion distributions on MRI. Typical MS showed ovoid lesions adjacent to the body of the lateral ventricles, Dawson’s fingers (periventricular demyelinating plaques along the axis), and T1 hypointense lesions, whereas fluffy lesions and three or less lesions were typical for MOGAD [40]. Lesion distribution in children seems to be age-dependent, with poorly demarcated, widespread lesions in younger children in contrast with a normal result in older children [41].
On orbital MRI, typical imaging of MOG-Ab-positive ON involve contrast enhancement of the optic nerve, perineural enhancement, and in 80%, more than half of the prechiasmic optic nerve length being affected, sparing the optic chiasm [42]. On spinal MRI, while patients with AQP4-Ab usually present cervical (with or without brainstem involvement) and thoracic lesions, MOG-Ab–positive patients can present lesions of the lower cord, including the conus medullaris [17,23]. Not all cases of MOG-Ab-positive TM are longitudinally extensive. A small proportion (7%) of MOG-Ab positivity were described to present with short myelitis occurring after an initial LETM episode or isolated at disease onset [30].
2. Cerebrospinal fluid results
Cerebrospinal fluid (CSF) reactivity to MOG has only been shown in patients with high serum levels [5,43]. Positivity for MOG-Ab in the CSF was found in 71% of patients who were MOG-Ab-seropositive, with a median CSF MOG-Ab titer of 1:4, lower than the serum titer [38]. IgG index, evidence of intrathecal synthesis, were generally absent [38]. These findings suggest a peripheral production of MOG-Ab and secondary diffusion in the CNS similar to that in NMOSD [44]. Findings of routine CSF analyses were comparable between MOGAD and NMOSD. CSF pleocytosis was detected in 55%–70%, with lymphocytic predominance [23,24], and cell counts higher than in typical MS [24,45]. Oligoclonal bands (OCBs) are uncommon (about 10%). Similarities were found between CSF cytokine profiles in MOGAD and AQP4-Ab–associated NMOSD, with a predominant up-regulation of T helper 17-related cytokines in the latter, whereas in MS, T helper 1-related cytokines were found [45].
3. Test for MOG antibodies
Several detection methods have been used to identify MOG-Ab. Given the inconsistent results generated by the enzyme-linked immunosorbent assay and immunoblot in MS patients, these techniques are not recommended [46]. Reliable results have been verified with cell-based assays, the detection of antibodies targeting human MOG using immunofluorescence or fluorescence-activated cell sorting. A recent review analyzed the differences in a summary of 26 studies [3]. A humoral response against MOG was predominantly present in ADEM with an overall sensitivity of 36.4%; it could also be detected in a substantial subgroup of patients with AQP4-negative ON, TM and NMOSD, with overall sensitivity of 26.9% [47].
A cutoff value is important, as low levels of MOG-Ab were also measured in healthy and other neurological controls. Most studies have used a cutoff of ≥1:160 [47]. A higher prognostic specificity was reported using a higher cutoff titer. The introduction of a cutoff at >1:1,280 MOG-Ab increased the specificity for non-MS disorders (96% 1:160 vs. 100% 1:1,280) or a recurrent non-MS courses (75% 1:160 vs. 86% 1:1,280). However, this higher cutoff at > 1:1,280 decreased the sensitivity for predicting non-MS disorders (from 47% to 31%) and a recurrent non-MS disease course (from 63% to 46%).
Prognosis
In several studies, MOG-Ab was associated with a favorable outcome [17]. MOG-Ab-positive patients less frequently suffer motor disability and have a better extended disability status scale (EDSS) score, compared to AQP4-Ab-positive patients [23]. Although, MOG-Ab-associated ON is usually a recurrent disease, visual recovery was good [42]; the outcome was better in MOG-than in AQP4-Ab-positive patients correlating with a better preserved retinal fiber layer thickness [42]. In patients with TM, the presence of MOG-Ab has also been associated with a better recovery from acute attacks, and really similar frequency of severe attacks at the onset to AQP4-Ab-associated disorders [19]. However, not all individuals will recover fully [17]. In a large cohort of MOG-Ab-positive patients followed for a median 28 months, 28% were left with permanent bladder dysfunction; 21% (among males) with erectile dysfunction; 20% with bowel dysfunction; 16% with decreased visual acuity in at least one eye; and 5% with an EDSS score ≥6 [31].
Monophasic and recurrent diseases are associated with MOG-Ab [48]. Older age, female sex, and MRI findings of atypical MS were risk factors for a recurrent disease course in MOGAD. The informed relapse varied across different studies. Relapse was observed in 36%–80% of patients, with an annualized relapse rate of 0.2–0.9, the highest risk in those with ON or NMOSD phenotypes [24,49]. These relapse rates seem to be lower than those of AQP4-positive patients [17,23,27]. These highly variable data of relapse rates may be due to the different characteristics of the enrolled patients among studies.
Diagnostic recommendations and algorithm for MOGADs
No clear distinctive phenotype has been identified in patients with MOG-Ab. The red flag signs and symptoms for the diagnosis of MOGAD are summarized in Table 2. Antibodies against MOG and AQP4 should be tested for in patients with clinical symptoms suggestive of NMOSD, such as bilateral ON, severe brainstem involvement, or LETM; in special patient groups with a high risk of NMOSD; if there is evidence of large cerebral lesions; if MS criteria of dissemination in space are not fulfilled; or if brain MRI findings are normal (Table 2) [50,51]. As a lack of OCBs is a very rare finding in typical MS, MOG antibody testing should be considered in OCB negative MS patients. In pediatric-onset MS, antibody testing can support the diagnosis of AQP4-negative NMOSD or ADEM followed by relapsing ON or including chronic relapsing inflammatory optic neuropathy. Other red flags indicating MOG-Ab testing include lesions with poorly demarcated margins located in the cerebellar peduncle and a leukodystrophy-like MRI pattern [51]. In 2017, Hacohen et al. precisely described the routine diagnostic use of MOG-Ab testing for pediatric patients in clinical practice and proposed a diagnostic algorithm for any episode of CNS demyelination [51]. In contrast, preceding criteria should include the McDonald criteria with caution for children under 12 years, as the validation of the predictive value is lacking [52].
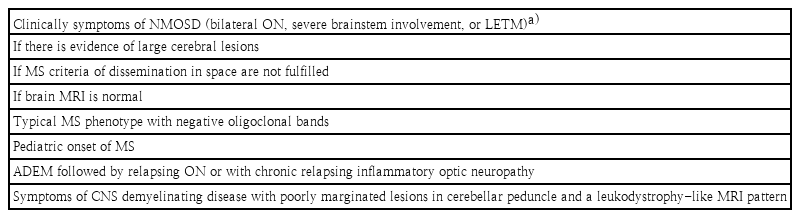
Diagnostic recommendations of MOG-Ab test for MOG-Ab-associated disorders
Di Pauli and Berger [50] generated a diagnostic algorithm and clini cal spectrum of MOGAD in patients with relapsing demyelinating syndromes (Fig. 1). As MS is the most common relapsing demyelinating disorder and reliably diagnosed with brain/spinal MRI and CSF study, brain/spinal MRI and CSF exams are recommended as a first step. In the case of typical MS findings, MS is diagnosed according to the revised McDonald criteria [53]. If, in clinically and/or radiologically suggested NMOSD, AQP4-Ab are negative, MOG-Ab should be tested. MOG-Ab testing is also recommended in patients with features of ADEM, particularly those with lesions with poorly demarcated margins located in the cerebellar peduncle or with a leukodystrophy-like MRI pattern. Physicians should be reminded that MOG-Ab-positive patients have distinct clinical features (young, less frequently affected by postrema area syndrome, typically presenting with ADEM initially, less disability during follow-up, and longer time to the first relapse [51]).

Diagnostic algorithm and clinical spectrum of MOG antibody-associated disorders. CNS, central nervous system; ON, optic neuritis; TM, transverse myelitis; MS, multiple sclerosis; ADEM, acute disseminated encephalomyelitis; CSF, cerebrospinal fluid; MRI, magnetic resonance imaging; LETM, longitudinally extensive transverse myelitis; MOG, myelin oligodendrocyte glycoprotein; IgG, immunoglobulin G; AQP4, aquaporin-4; NMOSD, neuromyelitis optica spectrum disorder.
Longitudinal analysis of MOG-antibodies: prognostic and clinical implications
Predicting the prognosis of MOG-Ab-positive patients considering the possibility of a multiphasic disease course has important suggestions concerning additional initiation of disease-modifying treatment. The correlation between longitudinal changes in antibody level and clinical progress has been evaluated [29,43]. Studies have discovered an association of MOG-Ab titer decrease with a monophasic course compared to stable or increasing titers in patients with multiphasic attacks [27,33]. Persistent MOG-Ab has been predominantly found in multiphasic ADEM, NMOSD, and ADEM followed by ON [54]. One study of adult and pediatric MOG-Ab-positive ADEM patients reinforced the usefulness of serial MOG-Ab testing for predicting relapse, as 88% of patients with persistent MOG-Ab relapsed during long-term follow-up versus 12% with transient antibodies [55]. These findings indicate that the maintenance of MOG-Ab–positivity could predict its clinical progress. On the other hand, Cobo-Calvo et al. [27] confirmed the trend toward an association between a relapsing disease and persistent antibodies only in a subgroup; in some groups, no such association was noticed. Similarly, Duignan et al. [56] found persistent MOG-Ab in relapsing and monophasic diseases alike. Long-term studies are needed to evaluate the usefulness of MOG-Ab as a biomarker and follow up the outcomes of demyelinating episodes in the developing CNS.
The conversion to seronegativity of MOG-Ab during immune therapies was a predictive marker for disease-free activity during the subsequent disease course [57]. However, more prospective data are needed to confirm the predictive value of serial MOG-Ab testing because the results are in part inconsistent and severe relapsing disease courses have been described in patients with decreasing/disappearing antibody levels.
Treatment
In an acute attack, similar strategies are used as in other CNS inflammatory or immune-mediated demyelinating diseases (intravenous methylprednisolone, plasma exchange, and intravenous immunoglobulin). A favorable recovery was demonstrated in 70%–90% of patients given pulsed intravenous methylprednisolone (dose 20–30 mg/kg/day for children, 500 mg to 1 g/day for adults over 3–5 days [17,58]). Patients were steroid responsive, but 70% of episodes treated with oral prednisone relapsed, particularly at doses <10 mg daily or within 2 months of cessation [58]. Long-term treatment with corticosteroids reduces the risk of relapse, and cessation has been associated with breakthrough disease [58]. Disease worsening or early relapse was observed after corticosteroid tapering or finishing [27,58]. Thus, some authors favored long-term oral prednisone treatment over 6 months (starting dose 1–2 mg/kg/day in children, 1 mg/kg in adults) given alongside other immune-modulating drugs. Use of plasma exchange seems to be associated with improved neurological deficits after corticosteroid failure [33].
A challenge in MOGAD is the choice of long-term immunotherapy since the pharmacological options for NMOSD have not been precisely evaluated in patients with MOGAD. Truly, it remains uncertain whether all MOGAD patients will require long-term treatment given the possibility of a mono phasic course, the commonly lower relapse rate, and the good recovery after attacks in response to acute treatment. A combination of corticosteroids and other immune-mediated therapies seems favorable. Some studies found a reduction in the annualized relapse rate with different immunotherapies such as azathioprine, rituximab, methotrexate, and mycophenolate, maintenance corticosteroids and rituximab being most effective at preventing disease activity, particularly if treatment is maintained for more than 3 months [27,58]. A recent study including children with relapsing MOGAD demonstrated a benefit of intravenous immunoglobulins on the annualized relapse rate [39]. It remains to be determined whether the disease course (i.e., single attack versus further relapses) or higher titers or persistence of MOG-Ab can predict the need for long-term therapy [16].
Typical MS drugs such as natalizumab, interferon, and glatiramer acetate revealed no treatment efficacy [24]. The failure of treatment efficacy in MS is well-known in NMOSD, suggesting a distinct pathomechanism in antibody-associated disorders.
Conclusion
Inflammatory demyelinating CNS diseases include a broad spectrum of diverse diseases, among which single diseases might show distinct clinical phenotypes and prognoses. Over the past few years, our knowledge of clinical, imaging, and laboratory data regarding MOGAD has developed. Despite some overlap, MOGAD exhibits different pathophysiological and phenotypic findings than both AQP4-Ab-associated NMOSD and typical MS. A different immunopathogenesis was found in MOG-Ab-associated NMOSD, with an oligodendrogliopathy rather than a classical astrocytopathy. This finding is clearly different from AQP4-associated disorders and suggests that other therapeutic strategies might be promising. MOG-Ab is of particular interest in pediatric patients with clinical or radiological non-MS typi cal findings and reliably predicts a non-MS demyelinating disease course. Indeed, early publications suggested that anti-MOG antibodies argue for a monophasic disease course, al though recent data have revealed an association with a risk for recurrence. Since persistent reactivity to MOG indicates a recurrent course, serial testing is recommended. Due to the increasing role of antibody testing, MOG-Ab was included in a diagnostic algorithm for children recommending for the first time a stand ardized use in clinical practice except in cases of typical MS. Further large prospective studies on the pathophysiology of MOGAD and the role of longitudinal analysis of antibodies are required to enable the development of evidence-based treatment strategies and assess the possible correlation of titers with disease activity.
Notes
No potential conflict of interest relevant to this article was reported.