Liver fibrosis in children: a comprehensive review of mechanisms, diagnosis, and therapy
Article information

Abstract
Chronic liver disease incidence is increasing among children worldwide due to a multitude of epidemiological changes. Most of these chronic insults to the pediatric liver progress to fibrosis and cirrhosis to different degrees. Liver and immune physiology differs significantly in children from adults. Because most of pediatric liver diseases have no definitive therapy, a better understanding of population and disease-specific fibrogenesis is mandatory. Furthermore, fibrosis development has prognostic significance and often guide treatment. Evaluation of liver fibrosis continues to rely on the gold-standard liver biopsy. However, many high-quality studies put forward the high diagnostic accuracy of numerous diagnostic modalities in this setting. Herein, we summarize and discuss the recent literature on fibrogenesis with an emphasis on pediatric physiology along with a detailed outline of disease-specific signatures, noninvasive diagnostic modalities, and the potential for antifibrotic therapies.
Key message
· Chronic liver diseases in children are heterogenous but converge in the common pathway of fibrosis.
· Much of the literature on mechanisms of fibrogenesis focus on adults but pediatric physiology has documented differences.
· Understanding of these distinctions are necessary to define, treat, and prevent fibrosis.
· Current management of liver fibrosis relies heavily on liver biopsy. Multiple tools have shown high diagnostic performance in pediatric and adult populations. Large, multicenter studies are needed for validation.

Graphical abstract.
HSC, hepatic stellate cell; ECM, excess extracellular matrix; ROS, reactive oxygen species; ER, endoplasmic reticulum; IFALD, intestinal failure-associated liver disease; NAFLD-NASH, nonalcoholic fatty liver disease-nonalcoholic steatohepatitis; AIH, autoimmune hepatitis; CHF, congestive heart failure; METAVIR, metaanalysis of histological data in viral hepatitis; VCTE, vibration-controlled transient elastography; USG, ultrasonography; MR, magnetic resonance; FXR, farnesoid X receptor; NK, natural killer; TIMP, tissue inhibitors for metalloproteinases.
Introduction
The incidence of chronic liver disease (CLD) is increasing among children worldwide owing to a multitude of epidemiological changes, such as the opioid epidemic for hepatitis C infection and obesity epidemic for nonalcoholic fatty liver disease (NAFLD) [1]. The overall incidence of liver disease in infants is 1 in 2,500, while in 2021, children represented 13% of all liver transplantations (LTs) in the US [2,3]. CLDs in pediatrics are heterogeneous and span a wide range of pathologies ranging from congenital or metabolic disorders to autoimmune diseases and viral diseases. Most chronic insults to the pediatric liver have the potential to progress to fibrosis and cirrhosis to different degrees.
The pathophysiology of fibrosis is a well-researched area with significant focus on novel biomarkers and antifibrotic treatments, especially in the adult population. Little is known about how this pathophysiology differs in the pediatric population, which has a unique hepatic and immune milieu. Moreover, certain pathologies in the pediatric population are distinct and characterized by different rates of fibrosis progression. For instance, fibrosis requires months to occur in all patients but neonates [4].
Depending on the etiology, follow-up of liver fibrosis is often required to define treatment goals. However, liver biopsy remains the gold-standard tool for this purpose despite its limitations such as invasiveness, complications, and inadequate sampling. Therefore, research has focused on noninvasive modalities and potential predictive biomarkers, and a review of these efforts in the pediatric population is required.
This article will outline the distinctions between adult and pediatric liver fibrosis with specific emphasis on pathophysiology, individual pediatric diseases, diagnostic modalities, and the ongoing search for therapeutic strategies for liver fibrosis.
Mechanisms of liver fibrosis
Liver fibrosis involves replacement of the hepatic parenchyma by excess extracellular matrix (ECM) as part of a wound healing response to chronic hepatic or bile duct injury [5]. The deposition of fibrillar collagen (i.e., collagen I, collagen III, and fibronectin) and unusual ECM proteins (e.g., chondroitin-sulfate rich proteoglycans) results in altered liver microarchitecture. These structural changes impede the bidirectional flow of plasma between the sinusoidal lumen and hepatocytes, resulting in further hepatic compromise [6].
In the liver, the major event that drives fibrogenesis is the activation and differentiation of hepatic stellate cells (HSCs) into α-smooth muscle actin (α-SMA)–positive myofibroblasts (MFs) with high profibrogenic capacity. In the normal liver, HSCs reside in the subendothelial space of Disse, the interface between hepatocytes and sinusoids, and store fat and vitamin A [7]. In a 2013 monumental study, HSCs were the major source of MFs in HSC-marked Cre-transgenic mouse models of toxic, cholestatic, and fatty liver disease [8]. In addition to the central role of HSC in fibrosis, recent studies revealed that multiple molecular and cellular processes are involved in this dynamic process (Fig. 1). Although experimental data are scarce, the pathophysiological steps outlined above may differ in multiple ways in the pediatric population (Fig. 2).
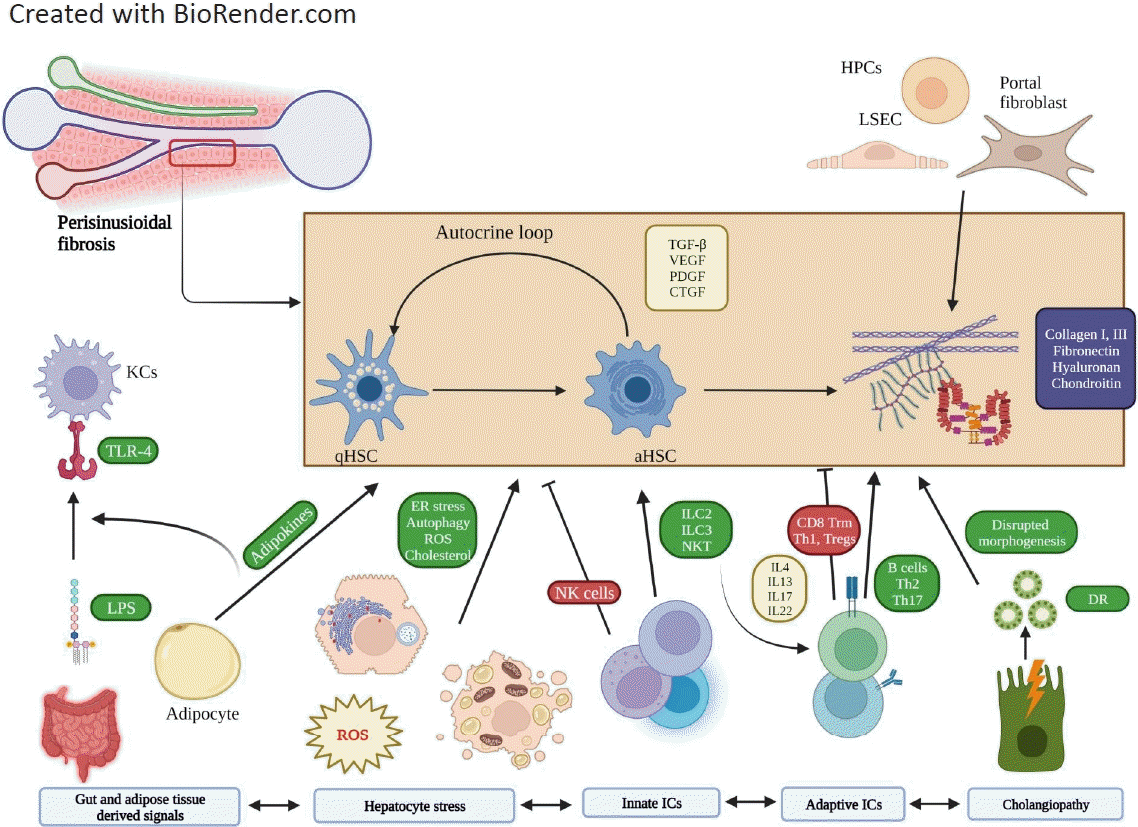
Cell-cell interactions and mediators driving hepatic stellate cell activation and fibrogenesis. Residing in the perisinusoidal space, HSCs become activated in response to a panoply of signals. LSECs, PFs, and HPCs also contribute to ECM deposition. Cytokines such as TGF-b, VEGF, PDGF, CTGF are implicated in promoting collagen production within an autocrine loop. Signals triggering this loop can be gut antigens acting on DAMP receptors on KCs, adipokines, products of cellular injury such as ROS, and some innate and adaptive immune cells. On the other hand, some immune cells participate in inhibition and removal of fibrosis. NK cells resident in the liver are antifibrotic through apoptosis of HSCs. Th1, Treg, and CD8 memory T cells also participate in fibrosis resolution. HSC, hepatic stellate cell; qHSC, quiescent HSC; aHSC, activated HSC; LSEC, liver sinusoidal endothelial cell; PF, portal fibroblast; HPC, hepatic progenitor cell; ECM, excess extracellular matrix; TGF-β, transforming growth factor-β; VEGF, vascular endothelial growth factor; PDGF, platelet-derived growth factor; CTGF, connective TGF; DAMP, damage-associated molecular pattern; KC, Kupffer cell; ROS, reactive oxygen species; NK, natural killer; Th1, T helper 1; Treg, regulatory T; TLR, toll-like receptor; LPS, lipopolysaccharide; ER, endoplasmic reticulum.
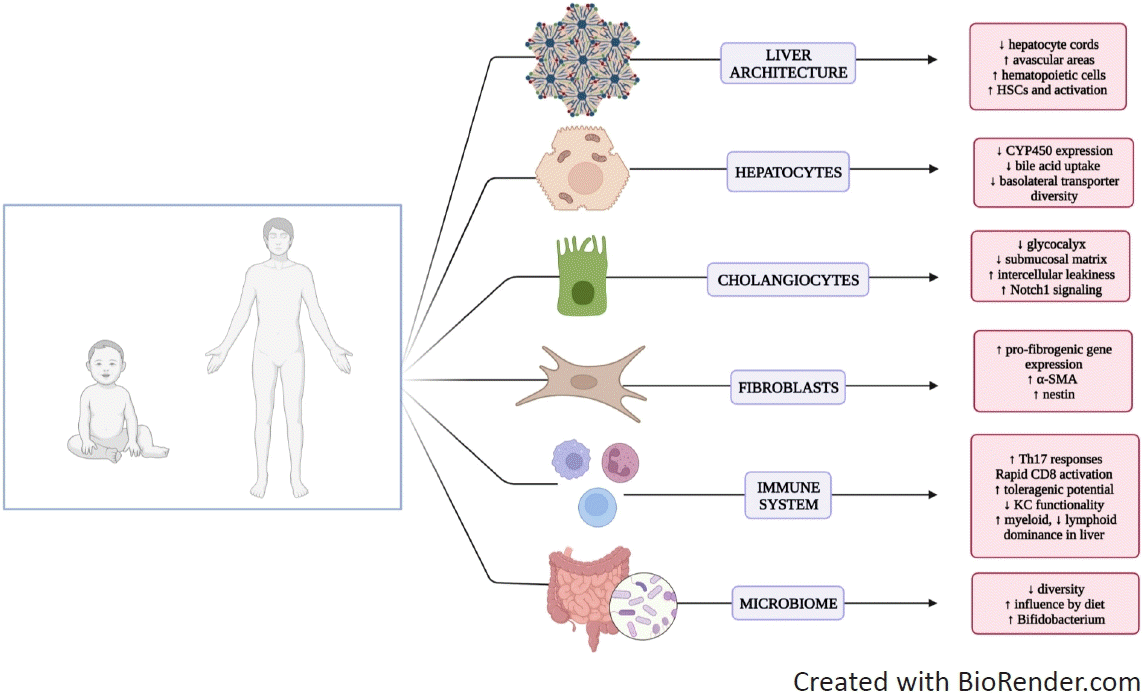
Distinct features of pediatric physiology that may contribute to the unique fibrosis pattern and progression seen in children. The process of neonatal or pediatric liver fibrosis differs from that in adults. Neonatal liver clearly lacks the mature architecture and enzyme levels. Similarly, cholangiocytes have immature barrier mechanisms that protect them from bile flow. Interestingly, the neonatal liver has a robust fibrogenic environment despite higher regenerative potential of the skin. The neonatal immune system is immature. CD8 T cells tend to differentiate into effector cells, while the hepatic immune system significantly differs in functionality and composition. The pediatric microbiome shifts across ages and impacts many disorders. CYP450, cytochromes P450; α-SMA, α-smooth muscle actin; Th, T helper; KC, Kupffer cell.
Pediatric liver diseases
Pediatric liver disease spans a large spectrum of diseases with heterogeneous pathophysiology, age range, and course. Liver diseases are hereditary except for NAFLD, viral hepatitis, primary sclerosing cholangitis (PSC), and autoimmune liver disease. Accordingly, the potential of these diseases to lead to fibrosis is different. Biliary atresia (BA), intestinal failure-associated liver disease (IFALD), and certain metabolic/genetic liver diseases characteristically begin in the neonatal period and manifest significant fibrosis early. Others, such as Alagille syndrome and autosomal recessive polycystic kidney disease, despite having a neonatal or even prenatal onset, present with fibrosis in older children. Table 1 summarizes the characteristics of chronic pediatric liver pathologies associated with fibrosis [6].

Frequency and characteristics of fibrosis development in chronic pediatric liver diseases
1. Cholestatic injury
1) Biliary atresia
BA is a progressive fibroinflammatory disease involving a segment or the entire extrahepatic biliary tree that leads to obliteration of the affected bile ducts within the first 3 months of life. To date, the exact pathophysiology of BA has remained elusive with a focus on environmental insults, immunologic dysregulation, genetic susceptibility, and developmental defects. The current hypothesis endorses a viral insult, leading to overactivation of the immune system. Fibrosis in BA is unique in that it progresses rapidly, leading to cirrhosis within a few weeks of birth in untreated cases. Kasai portoenterostomy (KPE) bypasses the atretic extrahepatic bile ducts and restores bile flow. The therapeutic outcome of KPE was evaluated using serum bilirubin levels at 6 months postoperative. Despite a good clinical response to KPE, liver fibrosis in BA often progresses to cirrhosis, requiring LT. In different series published worldwide, the rate of needing LT after KPE was 45%–70% at 5 years. Research has focused on identifying prognostic indicators in the post-KPE setting to predict progression to fibrosis and cirrhosis [9,10].
2) Primary sclerosing cholangitis
PSC is a chronic fibroinflammatory disorder of the intrahepatic and/or extrahepatic bile ducts of unknown etiology. The development of fibrosis is significant in pediatric PSC [11-16]. Despite the inconspicuous symptoms at presentation, such as fatigue and favorable magnetic resonance cholangiopancreatography scores, the majority of pediatric patients have significant fibrosis in the initial liver biopsy [17,18]. Around 1%–2% of adults, but 17%–30% of children progress to advanced liver disease and require LT each year [12,19]. In a large multicenter study of 781 children, Deneau et al. [11] found that a high fibrosis scoring aspartate transaminase (AST) to platelet ratio index (APRI) portends a poor prognosis.
No treatment options to date, such as oral vancomycin or ursodeoxycholic acid, are associated with improved fibrosis in pediatric PSC [20,21]. Haisma et al. [22] performed a genome-wide association study of 29 patient-parent trios with PSC onset at ≤12 years of age. They found pathogenic variants in 22 of 29 of patients and that some of the genes were involved in transmembrane transport, adaptive and innate immunity, and epithelial barrier function. On the other hand, adult genome studies identified most adult-onset susceptibility genes to the human leukocyte antigen complex II locus [23,24]. Therefore, pediatric PSC may represent a heterogeneous disease characterized by differing mechanisms and potential for fibrosis that may benefit from individualized treatment.
3) Alagille syndrome
Alagille syndrome is a multisystem disorder of morphogenesis that characteristically involves cholestasis, heart murmur, structural heart disease, dysmorphic facial features, vertebral anomalies, and optical findings. Through a mutation in the JAG1 gene on chromosome 20, commonly NOTCH2, the Notch signaling pathway is perturbed, resulting in complex developmental manifestations [25]. Liver involvement in Alagille syndrome is characterized by bile duct paucity, cholestasis, and fibrosis. Although the liver disease may improve with age, 43.5%–76% of patients reportedly require LT by adulthood [26-28].
Defective Notch signaling associated with Alagille syndrome has guided our understanding of biliary fibrosis. Upon injury, hepatic progenitor cells (HPCs) are activated and have the capacity to differentiate into hepatocyte as well as biliary epithelial cells in hepatocellular and biliary injury, respectively. Jagged-1 expressing portal MFs mediate the differentiation of HPCs upstream of Notch signaling. The outcome of Alagille syndrome is a scarcity of reactive ductular cells, resulting in thinner portal septa and an abundance of intermediate hepatic-biliary cells that promote intralobular chicken wire fibrosis [29,30].
2. Hepatocellular injuries
1) Chronic viral hepatitis
Most children acquire hepatitis B virus (HBV) and/or hepatitis C virus (HCV) infections in infancy, with 90% and 60% developing chronic infections, respectively [31,32]. Overall, chronic HCV infection in children follows an indolent course with low-level fibrosis. In 2 cohorts of 121 and 332 children, only 2 and 6 children developed cirrhosis in longitudinal follow-up, respectively [33,34]. Despite vaccination, HPV remains an important source of mortality and morbidity in children in the United States. In a study of histological specimens collected from 134 children, fibrosis and cirrhosis were present in 82% and 4%, respectively. In another study of 292 HBV-infected children, cirrhosis occurred in 3% at a median of 4 years of age [35,36].
In chronic HBV, Łotowska and Lebensztejn [37] found a positive correlation between the number of SMA-positive HSCs and fibrosis stage. The same group performed an ultrastructural phenotype analysis of chronic HBV biopsies and documented shifts in 2 cell types in children with advanced fibrosis: quiescent HSCs were replaced with transitional and MF-like HSCs and damaged liver progenitor cells in close proximity to the HSCs [38-40]. Close monitoring of fibrosis progression guides clinical decisions to begin antiviral therapy in these patients to prevent ongoing fibrosis [41].
2) Autoimmune hepatitis
Autoimmune hepatitis (AIH) is a chronic inflammatory disease of the liver characterized by hypergammaglobulinemia and autoantibody positivity. AIH is an important consideration in childhood liver fibrosis since one-third of patients present with cirrhosis and its complications [42,43]. Maia et al. [44] performed immunohistochemical examinations of pediatric AIH liver biopsies and noted that the activation of HSC populations decreased significantly following therapy. Studies involving follow-up biopsies in 16 and 13 pediatric patients with AIH demonstrated regression of fibrosis over 6–12 months [45,46].
3. Metabolic liver diseases
1) NAFLD–nonalcoholic steatohepatitis
NAFLD is a disease spectrum that ranges from hepatocyte steatosis to nonalcoholic steatohepatitis (NASH) to fibrosis and ultimately cirrhosis [47]. Fibrosis in NAFLD-NASH is perhaps the most studied pathophysiology in terms of mechanisms, diagnostics, and therapeutics. Atsawarungruangkit et al. [48] performed transient elastography-based screenings of 740 US adolescents embedded within the 2017–2018 National Health and Nutrition Examination Survey and identified advanced fibrosis secondary to NAFLD in 2.84%. According to an evaluation of liver biopsies from 100 children with NAFLD, 2 phenotypes of pediatric NASH exist: type I, with lobular inflammation and perisinusoidal fibrosis (17% of biopsies); and type II, with portal inflammation and fibrosis (51% of biopsies) [49]. Notably, children with the type II phenotype had more significant fibrosis.
The factors involved in driving the progression of NAFLD to fibrosis have been extensively evaluated. The 2 consistent risk factors for fibrosis development are metabolic syndrome and waist circumference [50-52]. Genetic variation is another source of wide study. Recently, Tao et al. performed a meta-analysis of 42 studies (>1 million participants), including 10 pediatric studies, to assess the role of rs641738C>T in MBOAT7. Although the variant was associated with advanced fibrosis on biopsy, its functional implications remain unclear. Another widely established variant is the rs738409 polymorphism in PNPLA3/adiponutrin. A meta-analysis of 5 studies with histological fibrosis assessments showed a significant association between advanced fibrosis and the variant [53]. In a study of 54 patients with NAFLD, those carrying the PNPLA3 variant had more periportal/portal fibrosis, hepatic progenitor cell activation, and serum oxidative stress [54]. Whether these findings can be applied to other etiologies could pose a worthwhile research question.
Environmental influences, such as dysbiosis and in utero exposure to a maternal Western diet, are related not only to NAFLD, but also to early liver fibrosis. NASH et al. recently demonstrated that nonhuman primate fetuses exposed to a maternal Western-style diet displayed increased fibrillar collagen deposition in the liver periportal region with localized HSC and MF activation. In a similar model, Friedman et al. [55] prevented collagen fibril formation in the offspring of mice fed a Westernstyle diet through a potent antioxidant, pyrroloquinoline quinone, which reversed macrophage metabolic programming from oxidative to glycolytic pathways and improved gut tight junction expression. Therefore, dietary modulation may be an important intervention for all patients with liver diseases.
2) Intestinal failure-associated liver disease
IFALD encompasses a spectrum of liver diseases, including steatosis, cholestasis, and fibrosis, in patients with intestinal failure and long-term use of parenteral nutrition (PN) [56]. It reportedly affects 22%–30% of such children according to clinical diagnoses [57,58]. However, in a recent study, surveillance biopsies in children receiving PN revealed an abnormal histology in 94% and 77% during and after PN, respectively. Of them, 88% and 64% had portal/periportal fibrosis [59]. The causes of IFALD are still being delineated with potential culprits involving plant sterols in PN lipid emulsion, gut dysbiosis, impaired barrier function, and episodes of sepsis. The fibrotic liver milieu in IFALD, even after weaning from PN, has been characterized by α-SMA+HSCs, increased collagen expression, and profibrotic/inflammatory cytokines, such as interleukin (IL)1a, IL1b, epithelial growth factor, adhesion molecule integrin-β6, and matrix metalloprotein (MMP)9 [60-62].
4. Systemic diseases
1) Congenital hepatic fibrosis
Congenital hepatic fibrosis is mostly associated with autosomal recessive polycystic kidney disease and a PKHD1 gene mutation that encodes for the transmembrane protein fibrocystin/polyductin, is localized to cilia, and is involved in the development of the renal collecting duct and biliary system [63]. Therefore, the characteristic pathological lesion in the liver is a ductal plate malformation, in which irregular biliary channels persist around the portal tracts [64]. Fibrosis can be severe, forming thick white bands of fibrous tissue across hepatocyte cell islands [65]. Mutation type affecting the PKHD1 gene determines the severity of associated polycystic kidney disease but not congenital hepatic fibrosis [66]. LT outcomes are promising, with good kidney and liver function, even in related living donor LT [67,68].
2) Cystic fibrosis liver disease
Cystic fibrosis (CF) liver disease (CFLD), which develops in 5–10% of patients with CF, is characterized by progressive hepatobiliary fibrosis. In a recent cohort study of 3,328 CF patients, of whom 32.2% and 10% developed CFLD and severe CFLD by 30 years of age, respectively [69]. Lewindon et al. [70] longitudinally followed 41 patients with CF and found that fibrosis at baseline biopsies, but not clinical or imaging abnormalities, heralded the development of clinically significant CFLD/portal hypertension.
However, the patchy nature of disease sampling may be inadequate for quantifying fibrosis. For the noninvasive diagnosis of liver fibrosis, Pereira et al. [71] correlated various serum markers of fibrosis in a cohort of 36 children with CFLD. Lewindon et al. [72] investigated the diagnostic performance of vibration-controlled transient elastography (VCTE) and APRI/Fibrosis-4 (FIB-4) Index for staging fibrosis in 27 and 51 children, respectively. In these studies, VCTE (area under the receiver operating characteristic curve [AUROC], 0.89), APRI (AUROC, 0.83), and prolyl hydroxylase (AUROC, 0.81) were superior to others in determining a meta-analysis of histological data in viral hepatitis (METAVIR) score ≥ F3 and Scheuer stage ≥2 fibrosis [72,73]. Future studies should provide clinical benefits to this patient group owing to the silent progression of fibrosis.
3) Fontan-associated liver disease
Fontan-associated liver disease (FALD) represents a congestive hepatopathy related to elevated central venous pressures that follow the Fontan operation and final-stage surgical palliation for univentricular physiology through complete cavopulmonary shunt creation [74]. According to studies on surveillance biopsies, nearly all biopsied patients develop significant fibrosis during adolescence [75]. Among our cohort of Fontan patients, FALD was detected in 87.5%. Liver stiffness showed a significant positive correlation with post-Fontan duration (P<0.05, r=0.550) and the FIB-4 index (r=0.7, P<0.05) but not with APRI. Splenomegaly (P<0.01, r=0.669), APRI (P<0.01, r=0.767), and FIB-4 index (P<0.01, r=0.922) were all correlated with postFontan duration, implying the importance of universal screening depending on the timing after surgery.
4) Fibrosis in LT
Owing to the improvements in post-LT care, the 10-year patient and graft survival rates in pediatric LT are 83% and 73%, respectively [76]. However, progressive liver fibrosis is among the most common causes of liver allograft failure in the pediatric group, reaching as high as 69%–97% according to different centers [77-81]. Both alloimmune inflammation and biliary outflow obstruction contribute to ongoing fibrosis [5]. Alterations in the gut microbiome and intestinal permeability are potential contributors through pathogen-associated molecular pattern translocation and pattern recognition receptor activation, abnormal bile acid metabolism, and short-chain fatty acid imbalances [82]. According to sequential allograft biopsies in the study by Varma et al. [83], the histological lesion begins with portal inflammation and progresses to periportal fibrosis, followed by extension to adjacent portal fields. Small longitudinal studies have shown that intensification of immunosuppression after histological diagnosis of inflammation and fibrosis leads to a reduction in fibrosis scores [80,84]. To date, studies investigating noninvasive techniques for identifying liver fibrosis in post-LT settings found that VCTE was superior to acoustic radiation force impulse (ARFI) and serum biomarkers (e.g., AST/alanine transaminase [ALT] ratio, APRI, FibroTest [FT], and enhanced liver fibrosis test) [85].
Approach to diagnosis
1. Liver biopsy
Liver biopsy is the ultimate tool used to evaluate liver fibrosis. However, a liver biopsy is often complicated by its cumbersome nature, inadequate sampling, and extended sampling time. Often, the fibrosis distribution is heterogeneous and may evolve quickly, such as in BA [86]. Studies of HCV-infected adults showed that fibrosis scoring varied depending on sampling site in 33%–45% of patients [87,88].
Fibrosis scoring systems facilitate the standardization and comparison of biopsies. To date, studies in children have utilized different scoring systems developed for evaluating fibrosis in adults and specific etiologies. These include the METAVIR (developed for hepatitis C scoring in adults), Ishak, Batts and Ludwig, Brunt, and Knodell classifications. The METAVIR as the most commonly used according to our review of the literature (68% of studies comparing biopsy grades with noninvasive tools). Each scoring system relies on the progressive development of periportal fibrosis. However, fibrosis in children may assume other patterns, such as chicken wire fibrosis within lobules, perisinusoidal involvement, patchy fibrosis, and more rapid development as in BA [89,90]. Therefore, diagnostic efforts have concentrated on the identification and validation of noninvasive and accurate tools for evaluating fibrosis.
2. Radiological assessment
Various radiological methods and their respective advantages and disadvantages are depicted in Table 2.
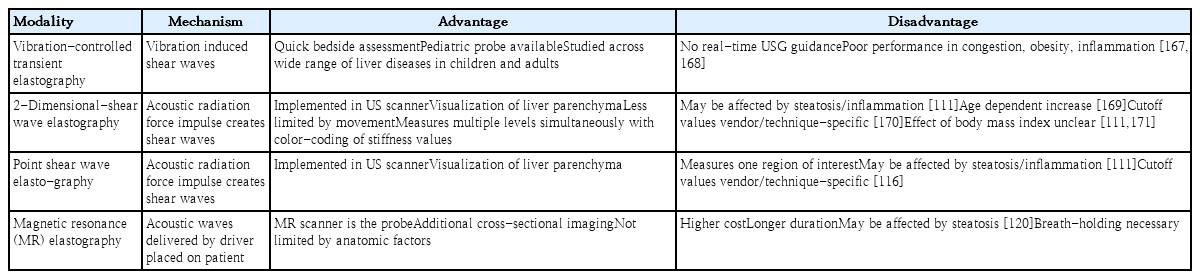
Radiological assessment tools for liver fibrosis
1) Vibration-controlled transient elastography
VCTE is the most commonly used modality for evaluating liver stiffness worldwide, with FibroScan (Echosens, Paris, France) being the most common product [91]. VCTE requires its own probe and relies on creating 50-Hz frequency vibrations to induce shear waves. The velocity at which the generated shear waves travel is directly related to liver stiffness and increases in stiffer tissue [92].
VCTE is the most well-studied modality for estimating liver fibrosis in adult and pediatric settings. In 2018, Hwang et al. [93] performed a meta-analysis of 11 studies involving 723 patients to investigate the correlation between liver fibrosis biopsies and VCTE measurements. The AUROC for detecting stage ≥F2 fibrosis was 0.96, which remained stable across subgroup analyses involving patients aged ≥8 years and study design.
Despite the high accuracy and reproducibility of liver stiffness measurements (LSMs), limitations exist. Studies of healthy children found that LSMs were higher with increasing age and in boys [94,95]. In addition, LSM cutoffs vary by etiology. Studies of heterogeneous patient populations found a cutoff level of 7.5–13 kPa for detecting stage ≥ F3/F4 fibrosis [96-99]. In studies of infants with BA, before KPE, the cutoffs were 9.8–23.2 kPa [100-102]. Therefore, validation studies in large cohorts with subgroup analyses are needed to improve the reliability and accuracy of VCTE in clinical practice.
2) Two-dimensional shear wave elastography and point shear wave elastography
Similar to VCTE, 2-dimensional shear wave elastography (2D-SWE) and point shear wave elastography (p-SWE) use shear waves created by the liver parenchyma that travel with higher velocity in a stiffer medium. In contrast to VCTE, 2D-SWE and p-SWE use ARFI to induce shear waves. Both modalities employ a regular ultrasound probe to take measurements; however, 2D-SWE samples have a larger area than p-SWE samples [103].
A 2016 meta-analysis of 13 articles on 2D-SWE in the diagnosis of liver fibrosis in adults found high sensitivity and specificity of 2D-SWE across all stages of fibrosis [104]. In a meta-analysis of 12 studies of 2D-SWE accuracy in fibrosis in children, Kim et al. [105] computed a summary sensitivity, specificity, and AUROC of 81%, 91%, and 0.93 for detecting stage ≥ F2 fibrosis, respectively. Median cutoff values for detecting significant fibrosis as summarized by Jiang et al. [104] and Kim et al. [105] were 8.0 kPa and 9.4 kPa in adults and children, respectively.
Individual studies have evaluated the diagnostic performance of 2D-SWE in disease groups and patients with potential confounders. Dhyani et al. [106] and Galina et al. [107] compared the accuracy and cutoff values of 2D-SWE across hepatocellular and cholestatic disease groups. They both found higher LSM cutoff values for detecting fibrosis in cholestatic groups. Chen et al. [108] did not report a significant difference between LSM cutoffs in BA and non-BA cholestatic infants, supporting that cholestasis, but not BA alone, impacts LSM. Moreover, Tutar and Alhashmi et al. found that steatosis increases the LSM cutoff and reduces overall performance of 2D-SWE, while in their cohort of 68 patients with NAFLD Garcovich et al. reported AUROC values of 0.92 and 0.97 for detecting stage ≥F1 and ≥F2 fibrosis, respectively [109-111]. Dardanelli et al. [112] identified hemosiderosis as a confounder of 2D-SWE.
Overall, p-SWE has been described as a tool with greater reliability than transient elastography but lower accuracy than 2D-SWE [113]. In their 2016 systematic review, Andersen et al. [114] included 6 studies that evaluated p-SWE versus liver biopsy performance. They found poor distinctive ability for no, mild, and moderate fibrosis and evidence of confounding by necroinflammatory activity. Our search revealed 3 additional publications with AUROC values of 0.83–0.86 for detecting METAVIR scores ≥F3 with cutoffs. Across all studies, the cutoff for detecting F3 fibrosis was 2.0–2.17 m/sec [115-117].
3) Magnetic resonance elastography
Similar to US-based techniques, magnetic resonance elastography (MRE) involves the creation and measurement of shear wave velocity that travels through the liver parenchyma. Propagating shear waves were measured using standard MR phase-contrast imaging sequences with added motion encoding gradients [118]. Performance statistics in previous adult studies were very promising as they found AUROC values of 0.83–0.99 and 0.84–1.0 for detecting stage ≥F2 and ≥F3 fibrosis, respectively [119]. To date, 3 large studies have evaluated the role of MRE in staging liver fibrosis, with AUROC values of 0.53–0.93 [120-122]. All studies were relatively uniform in terms of the patient population (age 13–14.2 years) and disease (mainly hepatocellular pattern). However, the accuracy and reliability of MRE in NAFLD was conflicting between the Trout and Schwimmer et al. studies, with AUROC values of 0.53 and 0.93 for this population, respectively.
3. Scores
Table 3 provides a brief overview of the studies that evaluated composite scores in pediatric settings.

Composite fibrosis scores evaluated in children with liver diseases (see Supplementary Table for details)
1) APRI and FIB-4
The APRI and FIB-4 scores were initially developed to monitor liver fibrosis progression in adults with hepatitis C [123,124]. Both are calculated using basic biochemistry, making them more accessible and cost-effective than radiological modalities.
The APRI was calculated using serum AST and platelet counts. The advancement of fibrosis reduces platelet counts in 2 ways: by reducing thrombopoietin generation by hepatocytes; and hypersplenism. Meanwhile, AST increases as chronic injury drives its release from mitochondria, and fibrosis diminishes clearance [125-127]. This pathophysiological derangement occurs later in the disease course, explaining why APRI has not always been a sensitive tool for detecting mild to moderate fibrosis [33,34]. Finally, APRI requires the use of the standard upper limit of normal for AST, which is 40 IU/L in most pediatric studies. Nevertheless, children often have lower baseline enzyme levels, which introduces potential bias to the score [128].
The FIB-4 index considers age-related differences in enzymes and ALT in addition to the components of APRI [123]. Despite these additional elements, APRI consistently outperformed FIB-4 in detecting fibrosis in both children and adults with liver disease [73,129-135]. One exception was the prediction of portal hypertension in a cohort of patients with CFLD, in which the FIB-4 index (AUROC, 0.91) had a superior AUROC to the APRI (AUROC, 0.71) [73].
2) FibroTest
FT is another composite score developed for the noninvasive evaluation of fibrosis in adults with chronic HCV. The FT combines age, sex, and 5 common serum markers: total bilirubin, a2-macroglobulin, haptoglobin, and gamma-glutamyl transferase [136]. Overall, the validation of FT in the pediatric cohort has been limited to relatively small studies comprising children with various CLD or viral hepatitis and did not reveal consistency [136-140].
3) Pediatric NAFLD Fibrosis Index and Pediatric NAFLD Fibrosis Score
Two indices were developed for noninvasive evaluation of liver fibrosis in children with NAFLD: Pediatric NAFLD Fibrosis Index (PNFI) and Pediatric NAFLD Fibrosis Score (PNFS). PNFI is based on age, waist circumference, and triglyceride levels. In a development cohort of 203 patients, the AUROC for predicting fibrosis was 0.85 [141]. Subsequently, Alkhouri et al. utilized PNFI in combination with an enhanced liver fibrosis test (ELF; including hyaluronic acid, procollagen-3, and tissue inhibitor of metalloproteinases-1) and VCTE. In both studies, ELF and VCTE were superior to PNFI, with AUROC values of 0.618–0.761 for detecting various fibrosis levels [142,143]. Similarly, Yang et al. [134] found that, across multiple diagnostic tests, PNFI had the lowest AUROC value in a cohort of 77 children with NAFLD. In 2014, Alkhouri et al. [131] developed PNFS in a cohort of 242 children with biopsy-proven NAFLD, which included ALT, alkaline phosphatase, platelet count, and gamma-glutamyl transferase levels. The AUROC value for this score was 0.74 for detecting stage ≥F3 fibrosis. However, external validation of this tool is still lacking [144].
4. Serum biomarkers
Serum biomarkers for liver fibrosis have historically been classified as direct or indirect. Direct markers reflect ECM turnover, while indirect markers are proteins that are altered in the serum due to hepatic dysfunction [145]. Table 4 summarizes the most widely studied biomarkers in the pediatric population.
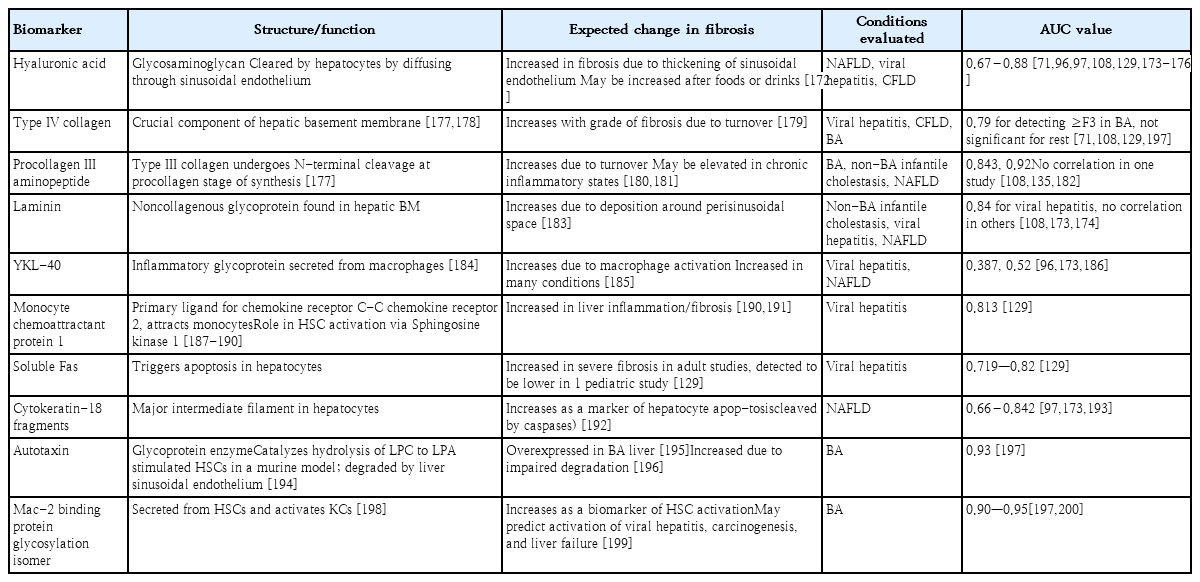
Most studied serum biomarkers of fibrosis in pediatric hepatology
Therapy
1. Fibrosis is reversible
Fibrosis was initially considered irreversible with permanent tissue scarring. However, there is now compelling evidence of its resolution in the liver. Patients with liver fibrosis due to AIH, Wilson disease, and viral hepatitis show its regression after adequate treatment of the underlying disease process [146]. This phenomenon hints at the inherent physiology of fibrosis turnover. The cessation of ongoing injury is key to resolving fibrosis. Subsequently, the hepatic immune milieu tips suppress inflammation and recruit restorative macrophages that express MMPs, growth factors, and phagocytosis-related receptors for ECM clearance [147]. With the loss of activating signals, MFs are targeted for apoptosis via natural killer and γδ T cell-mediated pathways [148].
2. Targeting ongoing liver injury
The resolution of fibrosis can interfere with ongoing injury. Unfortunately, despite progress in the molecular and genetic understanding of acquired and genetic liver disease in children, these conditions lack effective treatment except for a few such as viral hepatitis, autoimmune liver disease, and Wilson disease. Numerous trials investigating therapies directed at interfering with the disease process have incorporated liver fibrosis assessment as an endpoint. Several trials investigating the effect of metabolic modulation in NASH-NAFLD, such as through peroxisome proliferator-activated receptor-γ agonists (pioglitazone, rosiglitazone) on fibrosis regression, showed results ranging from decreased fibrosis to decreased fibrosis progression to no effect [149]. Similarly, ursodeoxycholic acid in primary biliary cirrhosis in a 4-year trial showed an overall improved rate of fibrosis progression, while a 2-year trial did not [149].
3. Antifibrotic therapies
Translational research on antifibrotic therapy has focused on targeting fibrosis and fibrolysis pathways [150]. Among the inhibitors of HSC activation (e.g., blockers of cannabinoid receptor 1 angiotensin-converting enzyme, angiotensin receptor, endothelin 1 receptor), the farnesoid X receptor (FXR) antagonist, obeticholic acid (OCA), has shown the greatest promise. FXR is strongly expressed in the liver and intestines and acts as a master transcriptional regulator of several enterohepatic metabolic pathways [151]. In a recent multicenter phase III study of 931 patients with F2–F3 fibrosis due to NASH, placebo, 10-mg OCA, and 25-mg OCA groups showed 12%, 18%, and 71% improvement rates in fibrosis [152]. Another approach is to eliminate HSCs through mechanisms that drive apoptosis. Selonsertib is a novel apoptosis signal-regulating kinase 1 inhibitor that promotes apoptosis and downregulates HSC activation. A phase II trial comparing 6 mg and 18 mg of selonsertib in patients with NASH fibrosis reported that 30% versus 43% of patients had improved fibrosis on repeat biopsies, but these results were not reproduced in trials involving patients with F3–F4 fibrosis [153,154]. Intracellularly acting to interfere with collagen production, pirfenidone has also demonstrated promising results in patients with HCV [155]. In this phase II trial, 67% of patients had improved fibrosis scores at the end of the 24-month period. In addition to pharmacological interventions, cellular therapies are being actively explored. In a novel in vivo study, engineered T cells programmed against a cardiac fibroblast activation protein demonstrated a significant reduction in cardiac fibrosis in mice [156].
Future directions and conclusion
The epidemiology and burden of CLD in children remains unknown. According to transplant registries and the World Health Organization, more than 500 children undergo LT, while approximately 30% die while awaiting LT [157,158]. As most pediatric liver diseases have no definitive therapy, a better understanding of disease-specific fibrogenesis is mandatory for this vulnerable population. Irrespective of the cause, a panoply of signals drives HSC activation. Additionally, crosstalk between liver sinusoidal endothelial cells, hepatic fibroblasts, macrophages, injured hepatocytes, and other cell types contributes to these mechanisms. The exact role of each cell type should be clarified by single-cell transcriptomic and proteomic studies.
Despite substantial progress and significant investment in the development of noninvasive markers of liver fibrosis, most have not yet been validated in children. Thus, there is an urgent need to develop effective and reliable noninvasive scoring systems to replace biopsies in children. In terms of pathological evaluation, digital pathology methods that leverage machine learning and artificial intelligence must be validated to obtain more prognostic information from liver biopsies.
In conclusion, liver fibrosis is a complex dynamic process involving parenchymal and nonparenchymal liver cells as well as immune cells. Further studies exploring cell-cell interactions and etiology-specific elements of fibrogenesis and their link to disease, as well as the mechanism of fibrosis resolution, are crucial to the development of more effective therapeutic targets.
Supplementary material
Supplementary Table can be found via https://doi.org/10.3345/cep.2022.00367.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.