About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 53(6); 2010
A case of Rubinstein-Taybi Syndrome with a CREB-binding protein gene mutation
- Corresponding Author: Jong Hee Chae, M.D. Seoul National University College of Medicine, Seoul National University Children's Hospital, 101 Daehangno, Jongno-gu, Seoul 110-744, Korea. Tel: +82.2-2072-3622, Fax: +82.2-743-3455, chaeped1@snu.ac.kr
- Received September 15, 2009 Revised October 08, 2009 Accepted October 27, 2009
- Abstract
-
Rubinstein-Taybi syndrome (RTS) is a congenital disorder characterized by typical facial features, broad thumbs and toes, with mental retardation. Additionally, tumors, keloids and various congenital anomalies including congenital heart defects have been reported in RTS patients. In about 50% of the patients, mutations in the CREB binding protein (CREBBP) have been found, which are understood to be associated with cell growth and proliferation. Here, we describe a typical RTS patient with Arnold-Chiari malformation. A mutation in the CREBBP gene, c.4944_4945insC, was identified by mutational analysis.
- Introduction
- Introduction
Rubinstein-Taybi syndrome (RTS) is a rare congenital disorder with distinctive facial appearance, broad thumbs, broad big toes and mental retardation. After the first report of the syndrome in 19631), various coexisting conditions such as talon cusp, congenital heart defects, or skin anomalies have been reported2). In 1991, a first sporadic case of RTS with de novo reciprocal translocation t(2:16)(p13.3;p13.3) was reported3), and a year later, mutation in the gene encoding CREB-binding protein (CREBBP) localized in 16p13.3 was identified to be the cause of RTS4). In approximately 50% of patients, mutations in the CREBBP gene have been found5). Although mutations in another EP300 gene have been reported in a small number of patients (3%)6), half of the clinically diagnosed RTS patients are still genetically not confirmed, suggesting RTS is genetically heterogeneous. RTS might have been unrecognized or underreported in Korea, because only a few cases are so far on record. Furthermore, mutation studies have never been reported in Korean RTS patients.Here, we report the first Korean case of genetically confirmed RTS associated with Arnold-Chiari malformation.
- Case report
- Case report
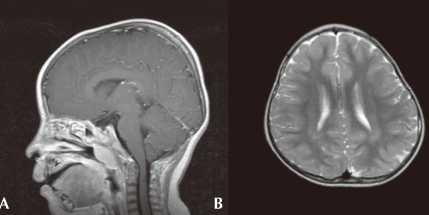
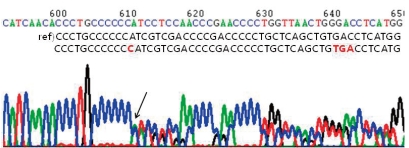
A four-year old boy first visited the outpatient clinic because of mental retardation and short stature. Born between a Korean father and a Japanese mother, the boy was delivered at term with a body weight of 3.68 kg without any perinatal problems. His initial crying was good, and his tone was normal. There was no other family member with mental retardation or neuromuscular disorder. He had received an operation on undescended testis when he was 8 months old. His developmental milestones were globally delayed. He controlled his head at 5 months, rolled over at 9 months, walked with assist at 17 months, and walked alone at 32 months. He could say only "mom" and "dad" at the age of 3. His height was 87.5 cm (<3 percentile), his weight 15.3 kg (10-25 p), and his head circumference 48.5 cm (3-50 p). Physical examination revealed a prominent forehead, highly arched eyebrows, down-slanted palpebral fissures, a broad nasal bridge, a short upper lip and a pouting lower lip, and a grimacing smile (Fig. 1A). Broad thumbs and terminal broadening of the phalanges of the fingers were also present (Fig. 1B). No focal neurologic signs were present, and his neurologic examination was normal.On ophthalmologic evaluation, an epiblepharon was revealed, but his fundus and disc were normal. Cardiac evaluation showed no abnormalities. His brain MRI study, taken when he was 3 years old, revealed multifocal lesions with increased signal intensity in the subcortical white matter of both temporal, left parietal regions and periventricular deep white matter (Fig. 2A). Arnold-Chiari malformation type I was also demonstrated (Fig. 2B). His chromosome study was normal with 46, XY and no abnormality was noted on tandem mass screening. His thyroid function test was normal.After obtaining informed consents from his parents, genetic analysis of his blood samples was performed. His genomic DNA was extracted from peripheral blood leukocytes using a Puregene DNA isolation kit (Gentra Systems Inc, Minneapolis, MN). Polymerase chain reaction was performed to amplify the entire coding and flanking regions of all exons of the CREBBP gene. Amplified products were sequenced bidirectionally using an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA). Mutation analysis demonstrated a CREBBP gene mutation with a C insertion in exon 31, nt 4944-4945 (Fig. 3).From the neuropsychologic evaluation (KEDI-WISC) done at the age of 6, his overall Intelligence quotient (IQ) was measured as 49, below normal. He scored low in both Performance IQ 55 and verbal IQ 53. He showed difficulties especially in fine-motor coordination and language skills. Special education with extra help was recommended for his school education.He received an operation on his epiblepharon when he was 6 years old, and he visits the outpatient clinic on a regular basis, and the six-year old boy is now able to speak sentences of 2-3 words.
- Discussion
- Discussion
RTS is a congenital disorder characterized by distinctive morphology. Typical facial features, big thumbs, big halluces, microcephaly, mental retardation, talon cusp and poor growth have been considered as the cardinal signs in RTS7). However in addition to these, many other anomalies in RTS have been reported including tumors, keloid tissues, congenital heart disease, genitourinary anomalies and joint abnormalities8). Premature thelarche9), and vascular ring10) have also been reported in RTS patients, although the relationship with the disease is not clear. Our patient also had typical features of facial dysmorphology, broad thumbs, mental retardation and cryptorchidism which is known to be associated with almost all males11). Additionally, Arnold-Chiari malformation type I and white matter abnormalities were identified on his brain magnetic resonance imaging (MRI) without any relevant symptoms. Although corpus callosal agenesis had been reported in a Brazillian boy with RTS12), only few studies have described the associated central nervous system (CNS) malformations or brain MRI findings12), including corpus callosal agenesis in a Brazillian boy with RTS13). Further studies would be meaningful to see whether the CNS abnormalities are also commonly associated findings in RTS patients.CREBPP is a transcriptional coactivator, that participates in cAMP regulated gene expression. It acts by binding to phosphorylated CREB, a transcription factor14), and is also thought to work with other transcription factors such as GLI3 in the Sonic Hedgehog-Patched-Gli (SHH) pathway15). It is not yet clear how exactly this protein causes multiple malformations with a broad spectrum of clinical features. However, it is understood to be involved in cell growth, transformation and development, and its intrinsic histone acetyltransferase (HAT) activity is considered to play a major role in the process.The CREBPP mutation in our patient, a frame shift mutation leading to early termination of transcription, was recently reported in a large group of patients being evaluated for genotype-phenotype correlations16). Although no significant correlations have been identified in previous studies17), this study suggests some correlation depending on the presence or type of the mutation: more frequent association with growth retardation in height and weight in patients with no CREBBP mutation, more frequent seizures in patients with CREBBP mutation, and a trend toward lower IQ and autistic features in patients with large deletions. Since genetic heterogeneity exist in RTS, careful genotype phenotype correlation would reveal separate groups that other specific genes might be involved. Therefore, further genetic tests may be deemed necessary, despite the fact that RTS could be diagnosed on clinical grounds.RTS is unique in that multiple congenital anomaly/intellectual impairment is caused by a generalized dysregulation of gene expression18). Recognizing these multiple disabilities or malformations by early diagnosis is important, because multidisciplinary approach is required in the initial evaluation, treatment, and follow-up. We hope, from the present study, more RTS patients would be recognized in the early course of the disease, and that the genetic and clinical characteristics of RTS in the Korean population would be further clarified.
- References
- 1. Rubinstein JH, Taybi H. Broad thumbs and toes and facial abnormalities. A possible mental retardation syndrome. Am J Dis Child 1963;105:588–608.
[Article] [PubMed]3. Imaizumi K, Kuroki Y. Rubinstein-Taybi syndrome with de novo reciprocal translocation t(2;16) (p13.3; p13.3). Am J Med Genet 1991;38:636–639.
[Article] [PubMed]4. Lacombe D, Saura R, Taine L, Battine J. Confirmation of assignment of a locus for Rubinstein-Taybi syndrome gene to 16p13.3. Am J Med Genet 1992;44:126–128.
[Article] [PubMed]5. Bentivegna A, Milani D, Gervasini C, Castronovo P, Mottadelli F, Manzini S, et al. Rubinstein-Taybi syndrome: spectrum of CREBBP mutations in Italian patients. BMC Med Genet 2006;7:77
[Article] [PubMed] [PMC]6. Roelfsema JH, White SJ, Ariyurek Y, Bartholldi D, Niedrist D, Papadia F, et al. Genetic heterogeneity in Rubinstein-Taybi syndrome: mutations in both the CBP and EP300 genes cause disease. Am J Hum Genet 2005;76:572–580.
[Article] [PubMed] [PMC]7. Wiley S, Swayne S, Rubinstein J, Lanphear N, Stevens CA. Rubinstein-Taybi syndrome medical guidelines. Am J Med Genet 2003;119A:101–110.
[Article] [PubMed]8. Roelfsema JH, Peters DJ. Rubinstein-Taybi syndrome: clinical and molecular overview. Expert Rev Mol Med 2007;9:1–16.
[Article]9. Kurosawa K, Masuno M, Imaizumi K, Matsuo M, Kuroki Y, Tachibana K. Premature thelarche in Rubinstein-Taybi syndrome. Am J Med Genet 2002;109:72–73.
[Article] [PubMed]10. Chun K, Colombani PM, Dudgeon DL, Haller JA. Diagnosis and management of congenital vascular rings: a 22-year experience. Ann Thorac Surg 1992;53:597–603.
[Article] [PubMed]11. Cantani A, Gagliesi D. Rubinstein-Taybi syndrome. Review of 732 cases and analysis of the typical traits. Eur Rev Med Pharmacol Sci 1998;2:81–87.
[PubMed]12. Sener RN. Rubinstein-Taybi syndrome: cranial MR imaging findings. Comput Med Imaging Graph 1995;19:417–418.
[Article] [PubMed]13. Guion-Almeida ML, Richieri-Costa A. Callosal agenesis, iris coloboma, and megacolon in a Brazilian boy with Rubinstein syndrome. Am J Med Genet 1992;43:929–931.
[Article] [PubMed]14. Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RCM, Masuno M, et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature 1995;376:348–351.
[Article] [PubMed]15. Villavicencio EH, Walterhouse DO, Iannaccone PM. The Sonic hedgehog-patched-gli pathway in human development and disease. Am J Hum Genet 2000;67:1047–1054.
[Article] [PubMed] [PMC]16. Schorry EK, Keddache M, Lanphear N, Rubinstein JH, Srodulski S, Fletcher D, et al. Genotype-phenotype correlations in Rubinstein-Taybi syndrome. Am J Med Genet 2008;146A:2512–2519.
[Article] [PubMed]
Fig. 1
Typcial facial features and broad thumbs of the patient. A) Note his small mouth, thin upper lip, micrognathia and antimongoloid slant of the palpebral fissures. B) Broad thumbs are noted in both hands.
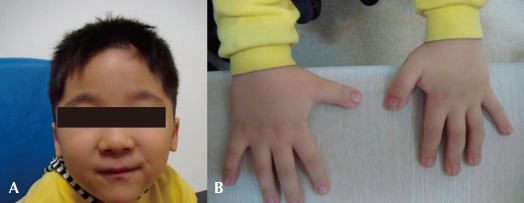
Fig. 2
Brain magnetic imaging of the patient. A) Sagittal T1-weighted image with gadolinium enhancement demonstrating mild downward displacement of cerebellar tonsil suggesting Arnold-Chiari malformation type I. B) Axial T2-weighted image demonstrating subtle high signal intensity of periventricular and deep white matter of bilateral hemisphere.