About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 53(7); 2010
Renal fibrosis
- Corresponding author: Min Hyun Cho, M.D. Department of Pediatrics, Kyungpook National University Hospital, 50, Samduk-dong 2-ga, Jung-gu, Daegu 700-721, Korea. Tel: +82.53-420-5719, Fax: +82.53-425-6683, chomh@knu.ac.kr
- Received May 19, 2010 Revised June 06, 2010 Accepted June 14, 2010
- Abstract
-
Renal fibrosis, characterized by tubulointerstitial fibrosis and glomerulosclerosis, is the final manifestation of chronic kidney disease. Renal fibrosis is characterized by an excessive accumulation and deposition of extracellular matrix components. This pathologic result usually originates from both underlying complicated cellular activities such as epithelial-to-mesenchymal transition, fibroblast activation, monocyte/macrophage infiltration, and cellular apoptosis and the activation of signaling molecules such as transforming growth factor beta and angiotensin II. However, because the pathogenesis of renal fibrosis is extremely complicated and our knowledge regarding this condition is still limited, further studies are needed.
- Introduction
- Introduction
Renal fibrosis, characterized by tubulointerstitial fibrosis and glomerulosclerosis, is the final manifestation of chronic kidney disease (CKD)1). The progression of CKD, one of the biggest problems in nephrology, indicates that patients inevitably reach end-stage renal disease (ESRD) and require renal replacement therapies such as dialysis and transplantation2). Although a range of diseases such as glomerulonephritis; metabolic diseases, including diabetes mellitus and atherosclerosis; obstructive nephropathy; interstitial nephritis; and cystic nephropathies, including polycystic kidney disease, can be the major causes of CKD, renal fibrosis is always the common ultimate result of CKD3, 4). Renal fibrosis is characterized by an excessive accumulation and deposition of extracellular matrix (ECM) components1). In order to elucidate the specific pathway of renal fibrosis, many experimental studies have been conducted using laboratory animals5-12). This review article will focus on recent advances in the pathogenesis of renal fibrosis and review the therapeutic trials conducted with an aim to overcome renal fibrosis.
- Experimental models of renal fibrosis
- Experimental models of renal fibrosis
Most of the present knowledge about the pathogenesis of renal fibrosis is based on experimental studies with laboratory animals. However, only few studies have reported the common features of renal fibrogenesis, such as interstitial fibrosis, tubular atrophy, and glomerulosclerosis5).Nephrotoxic serum nephritis (NSN) is a frequently used model for anti-glomerular basement membrane (anti-GBM) disease or in situ immune-complex glomerulonephritis and is characterized by an early heterologous phase, including linear deposition of anti-GBM antibody in the glomerulus and a subsequent autologous immune response against the planted antibodies6). NSN has been effectively used as a model for fibrosis with CD-1 mice, C57BL/6 mice, and various gene-knockout mice5). Fibrotic lesions, including collagen deposition in the interstitium, increased fibroblasts, epithelial-to-mesenchymal transition (EMT), appear after 1-2 weeks of NSN and severe tubulointerstitial fibrosis is noted between 3 and 6 weeks5).Mice with an intended disruption of the COL4A3 gene, which encodes the α3 chain of type IV collagen, were initially generated as a model for Alport syndrome7, 8). In this animal model, the primary renal pathologic finding is the splitting of the GBM and subsequent crescentic glomerulonephritis and renal fibrosis7, 9).Unilateral ureteral obstruction (UUO), as a model similar to human obstructive nephropathy, is induced by the ligation of a ureter of one kidney, while the contralateral kidney serves as a control; at as early as 3 days after UUO surgery, interstitial fibrosis associated with interstitial deposition of type IV collagen and tubular cell apoptosis are noted10). Thornhill et al.13) reported that tubular atrophy and interstitial fibrosis develop before significant renal pelvic dilatation in a neonatal rat model with variable chronic partial UUO, and that renal growth is reduced by 60% after 70% reduction of ureteral diameter. Moreover, although UUO is relieved after brief periods of obstruction, recovery from either structural or functional damages due to obstructive uropathy cannot be always guaranteed14).In the murine model of Denys-Drash syndrome, mice strains generated by crossbreeding of Wilms' tumor 1 gene (WT-1) knockout mice and mice with a yeast artificial chromosome containing the WT-1 locus can present with either crescentic glomerulonephritis or mesangial sclerosis depending on the relative WT-1 expression levels11). Assmann et al.12) reported that transgenic mice with ectopic expression of the Thy-1.1 antigen on the podocyte gradually and spontaneously develop focal glomerulosclerosis.
- Pathogenesis and therapeutic trials of renal fibrosis
- Pathogenesis and therapeutic trials of renal fibrosis
Renal fibrosis is characterized by glomerulosclerosis, tubulointerstitial fibrosis, loss of renal parenchyme, and inflammatory cell infiltration1) (Fig. 1A, 1B). These pathologic results usually originate from the underlying complicated cellular conditions such as the activation of EMT and fibroblasts, monocyte/macrophage infiltration, and cellular apoptosis1, 15). An early renal insult can evoke the activation of tubular cells that leads to the production of proinflammatory molecules that eventually contribute to renal fibrosis15). If high-grade proteinuria in tubular area develops due to the injured glomerular barrier, tubular cells can be exposed to bioactive molecules in the plasma or inflamed glomeruli16), thereby leading to the production of various chemotactic cytokines such as monocyte chemoattractant protein-1 (MCP-1)17, 18); regulated upon activation, normal T cell expressed and secreted (RANTES)19); and potent monocyte chemoattractants such as C3a and C5a20, 21). Moreover, leukocyte adhesion molecules such as osteopontin, intercellular adhesion molecules (ICAMs), and vascular cell adhesion molecules (VCAMs) have been reported to originate from tubules and to play an essential role in mononuclear cell recruitment in chronic renal disease state22, 23). Due to the activation of these various chemokines and chemoattractants, most of monocytes move into the glomerular and interstitial area from the circulation via peritubular capillary epithelium and infiltrated monocytes, leading to the production of inflammatory and fibrogenic cytokines, as well as injurious molecules, including reactive oxygen species (ROS)1). Finally, these inflammatory stimuli provoke the activation of mesangial cells, fibroblasts, and EMT and lead to the production of a large amount of ECM components. There are many disputes regarding the possible origins of renal fibroblasts that include migrating hematopoietic or mesenchymal stem cells from the bone marrow, periadventitial cells, activation of resident interstitial fibroblasts, and EMT of tubular epithelial cells24).In addition to this cellular activation, the activation of signal molecules also results in the accumulation of matrix along the tubular basement membranes and within the interstitial area15).Transforming growth factor beta (TGF-β) is regarded as the key mediator of renal fibrosis in CKD25). TGF-β can be produced by both resident kidney cells and infiltrating leukocytes and filtered from the plasma during proteinuria15). The 3 isoforms of TGF-β (TGF-β1, TGF-β2, and TGF-β3) are broadly expressed and act on almost every cell type in mammals by engaging in an intracellular signaling cascade of Smad family of proteins through ligand-induced activation of heteromeric transmembrane TGF-β receptor kinases25). In the UUO model, the downstream signaling of TGF-β1 is profoundly related to a family of Smad proteins that stimulate fibrosis (Smad2 and Smad3) or inhibit fibrosis (Smad7)26). TGF-β stimulates the myofibroblastic activation or transition of mesangial cells, interstitial fibroblasts, and tubular epithelial cells to become matrix-producing fibrogenic cells in vitro 1, 25). Overexpression of TGF-β also causes glomerular and interstitial fibrosis in transgenic mice27). These fibrotic effects of TGF-β can be summarized by 2 cellular events, apoptosis and EMT. TGF-β-induced apoptosis usually results in podocyte depletion, glomerulosclerosis, loss of glomerular and peritubular capillaries, and tubular atrophy28, 29). On the other hand, TGF-β-induced EMT can play a critical role in tubular atrophy and generation of interstitial myofibroblasts30). On the basis of this experimental background, many therapeutic trials on renal fibrosis such as neutralizing anti-TGF-β antibody and TGF-β receptor inhibitor have been performed31, 32). However, no conclusive results could be obtained since mice with TGF-β deficiency die of massive inflammation; this is because TGF-β is one of the anti-inflammatory cytokines. In addition, the over-expression of TGF-β1 in transgenic mice has been reported to attenuate the development of renal fibrosis via an anti-inflammatory effect33). Therefore, to improve the clinical application of TGF-β for preventing renal fibrosis, additional information regarding more complicated mechanisms underlying TGF-β action should be revealed.Angiotensin II (AngII) has also been known as one of the key mediators of inflammation and fibrosis in kidney diseases34). AngII signal transduction is initiated by 2 receptors, AngII type 1 (AT1) and AngII type 2 (AT2) receptors35). Most of the effects of AngII are mediated through the AT1 receptor, which is widely expressed by most cell types, whereas the expression of the AT2 receptor is higher in the fetal tissue and decreases in adult animals and humans36-38). Interestingly, the AT2 receptor has been thought to counteract the effects of AngII and to play a role in the protection of the kidney 39). AngII through AT1 receptor regulates ECM accumulation mediated by profibrotic growth factors such as TGF-β34). Clinically, in humans with chronic renal allograft rejection, plasma TGF-β levels were found to decrease after AngII was blocked40). AngII also stimulates oxidative stress, which in turn upregulates the vasoconstrictor peptides due to increased catabolism of nitric oxide (NO)41). Because NO plays an essential role in the regulation of blood flow in the normal and diseased kidneys, the changes of blood flow in UUO are associated with the impairment of the NO synthetic pathway in the kidney42). However, because angiotensin itself has a very important role in the development and maturation of a normal kidney, an angiotensin-converting enzyme (ACE) inhibitor can aggravate renal interstitial inflammation and fibrosis in a neonatal rat model with partial UUO43).Connective tissue growth factor (CTGF) is an important profibrotic factor that contributes to renal fibrosis and tubuloepithelial transdifferentiation as a downstream mediator of TGF-β activity44). Interstitial production of CTGF was found to increase in areas of chronic renal injury in humans45) and to be attenuated by ACE inhibitors and antagonists of AT1 receptor46, 47). Lin et alreported that pentoxifylline suppresses the activity of CTGF in rats with UUO by interfering with Smad3- and Smad4-dependent CTGF transcription48).Platelet-derived growth factor (PDGF) is also centrally involved in the pathogenesis of renal fibrosis, similar to CTGF3, 49). In a rat model with progressive mesangioproliferative glomerulonephritis, the treatment with an antagonist against PDGF-B attenuated mesangioproliferative changes, glomerular hypertrophy, and podocyte damage, and in the long term prevented glomerulosclerosis and tubulointerstitial damage49). Of the PDGF family, PDGF-C and D are also increased in the area of tubulointerstitial fibrotic lesion, suggesting a potential role in renal fibrosis50, 51).Endothelin-1 (ET-1) that is abundant in renal endothelial cells is a strong vasoconstrictor peptide with profibrotic and pro-inflammatory effects52). Glomerular ET-1 expression was remarkably elevated in streptozotocin-induced diabetic rats53). Overexpression of ET-1 is sufficient to induce structural and functional changes, including glomerular and tubulointerstitial fibrosis, because ET-1 causes hypoxic injury due to the constriction of peritubular capillaries and stimulates mesangial cell proliferation and ECM production54, 55). Kon et al.56) suggested that antagonism of ET-1 may attenuate tubulointerstitial injury in an experimental model of chronic cyclosporine nephrotoxicity.In addition, tumor necrosis factor alpha (TNF-α) and interleukin-1 (IL-1) are other candidate proinflammatory cytokines that can lead to renal fibrosis57, 58).Simultaneously, other antifibrotic factors may be involved in the pathway of renal fibrosis combined with profibrotic cytokines and growth factors.Hepatocyte growth factor (HGF), a promoter of hepatocyte proliferation and liver generation, is also regarded as an antifibrotic mediator of tubular repair and generation after acute renal injury59, 60). Moreover, HGF prevents the initiation and progression of chronic renal fibrosis and attenuates TGF-β1 expression in various animal models60). In a mice model with UUO, the administration of exogenous HGF noticeably reduced the progression of renal fibrosis and attenuated the expression of α-smooth muscle actin, TGF-β61). In addition, in an animal model of diabetic nephropathy, HGF interrupted the progression of renal fibrosis and dysfunction62). These antifibrotic results of HGF can be explained by various antagonistic effects on the cellular activation of renal fibrosis. HGF blocks tubular EMT63), inhibits apoptosis of endothelial cells64), and accelerates matrix degradation by modulating both the plasminogen activator/plasmin and matrix metalloproteinases (MMPs) proteolytic pathway65).Bone morphogenetic protein-7 (BMP-7), formerly known as osteogenic protein-1, is a member of the TGF-β superfamily and a key growth factor of embryogenesis and morphogenesis66). Recently, BMP-7 has been reported to reverse renal fibrosis induced by TGF-β in a mice model and to antagonize TGF-β-dependent fibrogenesis in mesangial cells67, 68).In addition, interferon gamma (IFN-γ) and insulin-like growth factor-1 (IGF-1) are also known to have an antifibrotic effect57, 69).There is always a balance between matrix production and degradation in the normal renal tissue, whereas the excessive accumulation and deposition of ECM components are the final pathologic results in renal fibrosis and CKD. Fibrotic appearance in renal fibrosis may be induced by both overproduction of matrix components and defects in their degradation1). The normal kidney usually produces various proteases, including MMP, plasminogen activator, and lysosomal cathepsins70). The balance between matrix production and degradation can be maintained through appropriate degradation of ECM due to the potent proteases. Therefore, these proteases have been regarded as one of the possible candidates for the treatment of renal fibrosis70). However, recent data suggest that the action mechanisms of proteases are not simple. In an animal model, ironically, deficiency of plasminogen attenuates renal fibrosis, and overproduction of MMP produces spontaneous EMT and peritubular fibrosis in renal proximal tubules71, 72). Fig. 2 summarizes the cellular and molecular activation of renal fibrosis (Fig. 2).Although we have secured various possible targets for the treatment of renal fibrosis as mentioned above, it is evident that there is no effective treatment for renal fibrosis thus far. Boor et al.73) reported several problems that even we faced, including discrepancy between rodent models and clinical situation, lack of multiple models, different parameters and techniques for the evaluation of fibrosis, and inadequate timing of treatment in the experimental fibrosis.
- Conclusion
- Conclusion
Although renal fibrosis, including tubulointerstitial fibrosis and glomerulosclerosis, is always the final common outcome in CKD, the pathogenic mechanisms underlying renal fibrosis are extremely complicated. Both cellular activation such as EMT; fibroblast activation; monocyte/macrophage infiltration; cellular apoptosis, and the activation of profibrotic cytokines such as TGF-β, AngII, CTGF, PDGF, ET-1, TNF-α, and IL-1 result in the excessive accumulation of ECM. Antifibrotic cytokines such as HGF, BMP-7, IFN-γ, and IGF-1 or several proteases such as MMP and plasminogen activator may be possible candidates for the treatment of renal fibrosis; however, our knowledge about these candidates is still limited. Further studies are needed to elucidate the complicated pathogenesis of renal fibrosis and to overcome the irreversibility of CKD.
- References
- 1. Liu Y. Renal fibrosis: new insights into the pathogenesis and therapeutics. Kidney Int 2006;69:213–217.
[Article] [PubMed]2. el Nahas AM, Muchaneta-Kubara EC, Essawy M, Soylemezoglu O. Renal fibrosis: insights into pathogenesis and treatment. Int J Biochem Cell Biol 1997;29:55–62.
[Article] [PubMed]3. Eitner F, Floege J. Novel insights into renal fibrosis. Curr Opin Nephrol Hypertens 2003;12:227–232.
[Article] [PubMed]4. Remuzzi G, Bertani T. Pathophysiology of progressive nephropathies. N Engl J Med 1998;339:1448–1456.
[Article] [PubMed]5. Zeisberg M, Soubasakos MA, Kalluri R. Animal models of renal fibrosis. Methods Mol Med 2005;117:261–272.
[PubMed]6. Lloyd CM, Minto AW, Dorf ME, Proudfoot A, Wells TN, Salant DJ, et al. RANTES and monocyte chemoattractant protein-1 (MCP-1) play an important role in the inflammatory phase of crescentic nephritis, but only MCP-1 is involved in crescent formation and interstitial fibrosis. J Exp Med 1997;185:1371–1380.
[Article] [PubMed] [PMC]7. Cosgrove D, Meehan DT, Grunkemeyer JA, Kornak JM, Sayers R, Hunter WJ, et al. Collagen COL4A3 knockout: a mouse model for autosomal Alport syndrome. Genes Dev 1996;10:2981–2992.
[Article] [PubMed]8. Miner JH, Sanes JR. Molecular and functional defects in kidneys of mice lacking collagen alpha 3(IV): implications for Alport syndrome. J Cell Biol 1996;135:1403–1413.
[Article] [PubMed] [PMC]9. Hamano Y, Grunkemeyer JA, Sudhakar A, Zeisberg M, Cosgrove D, Morello R, et al. Determinants of vascular permeability in the kidney glomerulus. J Biol Chem 2002;277:31154–31162.
[Article] [PubMed]10. Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 2002;110:341–350.
[Article] [PubMed] [PMC]11. Guo JK, Menke AL, Gubler MC, Clarke AR, Harrison D, Hammes A, et al. WT1 is a key regulator of podocyte function: reduced expression levels cause crescentic glomerulonephritis and mesangial sclerosis. Hum Mol Genet 2002;11:651–659.
[Article] [PubMed]12. Assmann KJ, van Son JP, Dijkman HB, Mentzel S, Wetzels JF. Antibody-induced albuminuria and accelerated focal glomerulosclerosis in the Thy-1.1 transgenic mouse. Kidney Int 2002;62:116–126.
[Article] [PubMed]13. Thornhill BA, Burt LE, Chen C, Forbes MS, Chevalier RL. Variable chronic partial ureteral obstruction in the neonatal rat: a new model of ureteropelvic junction obstruction. Kidney Int 2005;67:42–52.
[Article] [PubMed]14. Chevalier RL, Thornhill BA, Wolstenholme JT, Kim A. Unilateral ureteral obstruction in early development alters renal growth: dependence on the duration of obstruction. J Urol 1999;161:309–313.
[Article] [PubMed]16. Wang SN, Lapage J, Hirschberg R. Glomerular ultrafiltration and apical tubular action of IGF-I, TGF-beta, and HGF in nephrotic syndrome. Kidney Int 1999;56:1247–1251.
[Article] [PubMed]17. Wang Y, Chen J, Chen L, Tay YC, Rangan GK, Harris DC. Induction of monocyte chemoattractant protein-1 in proximal tubule cells by urinary protein. J Am Soc Nephrol 1997;8:1537–1545.
[Article] [PubMed]18. Grandaliano G, Gesualdo L, Ranieri E, Monno R, Montinaro V, Marra F, et al. Monocyte chemotactic peptide-1 expression in acute and chronic human nephritides: a pathogenetic role in interstitial monocytes recruitment. J Am Soc Nephrol 1996;7:906–913.
[Article] [PubMed]19. Lloyd CM, Dorf ME, Proudfoot A, Salant DJ, Gutierrez-Ramos JC. Role of MCP-1 and RANTES in inflammation and progression to fibrosis during murine crescentic nephritis. J Leukoc Biol 1997;62:676–680.
[Article] [PubMed]20. Biancone L, David S, Della Pietra V, Montrucchio G, Cambi V, Camussi G. Alternative pathway activation of complement by cultured human proximal tubular epithelial cells. Kidney Int 1994;45:451–460.
[Article] [PubMed]21. Tang S, Sheerin NS, Zhou W, Brown Z, Sacks SH. Apical proteins stimulate complement synthesis by cultured human proximal tubular epithelial cells. J Am Soc Nephrol 1999;10:69–76.
[Article] [PubMed]22. Ricardo SD, Levinson ME, DeJoseph MR, Diamond JR. Expression of adhesion molecules in rat renal cortex during experimental hydronephrosis. Kidney Int 1996;50:2002–2010.
[Article] [PubMed]23. Okada H, Moriwaki K, Kalluri R, Takenaka T, Imai H, Ban S, et al. Osteopontin expressed by renal tubular epithelium mediates interstitial monocyte infiltration in rats. Am J Physiol Renal Physiol 2000;278:F110–F121.
[Article] [PubMed]24. Strutz F, Muller GA. Renal fibrosis and the origin of the renal fibroblast. Nephrol Dial Transplant 2006;21:3368–3370.
[Article] [PubMed]25. Bottinger EP, Bitzer M. TGF-beta signaling in renal disease. J Am Soc Nephrol 2002;13:2600–2610.
[Article] [PubMed]26. Fukasawa H, Yamamoto T, Togawa A, Ohashi N, Fujigaki Y, Oda T, et al. Down-regulation of Smad7 expression by ubiquitin-dependent degradation contributes to renal fibrosis in obstructive nephropathy in mice. Proc Natl Acad Sci U S A 2004;101:8687–8692.
[Article] [PubMed] [PMC]27. Kopp JB, Factor VM, Mozes M, Nagy P, Sanderson N, Bottinger EP, et al. Transgenic mice with increased plasma levels of TGF-beta 1 develop progressive renal disease. Lab Invest 1996;74:991–1003.
[PubMed]28. Schuster N, Krieglstein K. Mechanisms of TGF-beta-mediated apoptosis. Cell Tissue Res 2002;307:1–14.
[Article] [PubMed]29. Schiffer M, Bitzer M, Roberts IS, Kopp JB, ten Dijke P, Mundel P, et al. Apoptosis in podocytes induced by TGF-beta and Smad7. J Clin Invest 2001;108:807–816.
[Article] [PubMed] [PMC]30. Oldfield MD, Bach LA, Forbes JM, Nikolic-Paterson D, McRobert A, Thallas V, et al. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). J Clin Invest 2001;108:1853–1863.
[Article] [PubMed] [PMC]31. Schnaper HW, Hayashida T, Hubchak SC, Poncelet AC. TGF-beta signal transduction and mesangial cell fibrogenesis. Am J Physiol Renal Physiol 2003;284:F243–F252.
[Article] [PubMed]32. Ma LJ, Jha S, Ling H, Pozzi A, Ledbetter S, Fogo AB. Divergent effects of low versus high dose anti-TGF-beta antibody in puromycin aminonucleoside nephropathy in rats. Kidney Int 2004;65:106–115.
[Article] [PubMed]33. Wang W, Huang XR, Li AG, Liu F, Li JH, Truong LD, et al. Signaling mechanism of TGF-beta1 in prevention of renal inflammation: role of Smad7. J Am Soc Nephrol 2005;16:1371–1383.
[Article] [PubMed]34. Ruiz-Ortega M, Ruperez M, Esteban V, Rodriguez-Vita J, Sanchez-Lopez E, Carvajal G, et al. Angiotensin II: a key factor in the inflammatory and fibrotic response in kidney diseases. Nephrol Dial Transplant 2006;21:16–20.
[Article] [PubMed]35. Yayama K, Okamoto H. Angiotensin II-induced vasodilation via type 2 receptor: role of bradykinin and nitric oxide. Int Immunopharmacol 2008;8:312–318.
[Article] [PubMed]36. Matsubara H. Pathophysiological role of angiotensin II type 2 receptor in cardiovascular and renal diseases. Circ Res 1998;83:1182–1191.
[Article] [PubMed]37. Viswanathan M, Tsutsumi K, Correa FM, Saavedra JM. Changes in expression of angiotensin receptor subtypes in the rat aorta during development. Biochem Biophys Res Commun 1991;179:1361–1367.
[Article] [PubMed]38. Batenburg WW, Garrelds IM, Bernasconi CC, Juillerat-Jeanneret L, van Kats JP, Saxena PR, et al. Angiotensin II type 2 receptor-mediated vasodilation in human coronary microarteries. Circulation 2004;109:2296–2301.
[Article] [PubMed]39. Wenzel UO, Krebs C, Benndorf R. The angiotensin II type 2 receptor in renal disease. J Renin Angiotensin Aldosterone Syst 2010;11:37–41.
[Article] [PubMed]40. Campistol JM, Inigo P, Jimenez W, Lario S, Clesca PH, Oppenheimer F, et al. Losartan decreases plasma levels of TGF-beta1 in transplant patients with chronic allograft nephropathy. Kidney Int 1999;56:714–719.
[Article] [PubMed]41. Warnholtz A, Nickenig G, Schulz E, Macharzina R, Brasen JH, Skatchkov M, et al. Increased NADH-oxidase-mediated superoxide production in the early stages of atherosclerosis: evidence for involvement of the renin-angiotensin system. Circulation 1999;99:2027–2033.
[Article] [PubMed]42. Hegarty NJ, Young LS, Kirwan CN, O'Neill AJ, Bouchier-Hayes DM, Sweeney P, et al. Nitric oxide in unilateral ureteral obstruction: effect on regional renal blood flow. Kidney Int 2001;59:1059–1065.
[Article] [PubMed]43. Chen CO, Park MH, Forbes MS, Thornhill BA, Kiley SC, Yoo KH, et al. Angiotensin-converting enzyme inhibition aggravates renal interstitial injury resulting from partial unilateral ureteral obstruction in the neonatal rat. Am J Physiol Renal Physiol 2007;292:F946–F955.
[Article] [PubMed]44. Perbal B. CCN proteins: multifunctional signalling regulators. Lancet 2004;363:62–64.
[Article] [PubMed]45. Ito Y, Aten J, Bende RJ, Oemar BS, Rabelink TJ, Weening JJ, et al. Expression of connective tissue growth factor in human renal fibrosis. Kidney Int 1998;53:853–861.
[Article] [PubMed]46. Ruperez M, Lorenzo O, Blanco-Colio LM, Esteban V, Egido J, Ruiz-Ortega M. Connective tissue growth factor is a mediator of angiotensin II-induced fibrosis. Circulation 2003;108:1499–1505.
[Article] [PubMed]47. Esteban V, Lorenzo O, Ruperez M, Suzuki Y, Mezzano S, Blanco J, et al. Angiotensin II, via AT1 and AT2 receptors and NF-kappaB pathway, regulates the inflammatory response in unilateral ureteral obstruction. J Am Soc Nephrol 2004;15:1514–1529.
[Article] [PubMed]48. Lin SL, Chen YM, Chien CT, Chiang WC, Tsai CC, Tsai TJ. Pentoxifylline attenuated the renal disease progression in rats with remnant kidney. J Am Soc Nephrol 2002;13:2916–2929.
[Article] [PubMed]49. Ostendorf T, Kunter U, Grone HJ, Bahlmann F, Kawachi H, Shimizu F, et al. Specific antagonism of PDGF prevents renal scarring in experimental glomerulonephritis. J Am Soc Nephrol 2001;12:909–918.
[Article] [PubMed]50. Eitner F, Ostendorf T, Van Roeyen C, Kitahara M, Li X, Aase K, et al. Expression of a novel PDGF isoform, PDGF-C, in normal and diseased rat kidney. J Am Soc Nephrol 2002;13:910–917.
[Article] [PubMed]51. Changsirikulchai S, Hudkins KL, Goodpaster TA, Volpone J, Topouzis S, Gilbertson DG, et al. Platelet-derived growth factor-D expression in developing and mature human kidneys. Kidney Int 2002;62:2043–2054.
[Article] [PubMed]52. Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988;332:411–415.
[Article] [PubMed]53. Hargrove GM, Dufresne J, Whiteside C, Muruve DA, Wong NC. Diabetes mellitus increases endothelin-1 gene transcription in rat kidney. Kidney Int 2000;58:1534–1545.
[Article] [PubMed]54. Richter CM. Role of endothelin in chronic renal failure--developments in renal involvement. Rheumatology (Oxford) 2006;45(Suppl 3): iii36–iii38.
[Article] [PubMed]55. Sorokin A, Kohan DE. Physiology and pathology of endothelin-1 in renal mesangium. Am J Physiol Renal Physiol 2003;285:F579–F589.
[Article] [PubMed]56. Kon V, Hunley TE, Fogo A. Combined antagonism of endothelin A/B receptors links endothelin to vasoconstriction whereas angiotensin II effects fibrosis. Studies in chronic cyclosporine nephrotoxicity in rats. Transplantation 1995;60:89–95.
[Article] [PubMed]57. Guo G, Morrissey J, McCracken R, Tolley T, Klahr S. Role of TNFR1 and TNFR2 receptors in tubulointerstitial fibrosis of obstructive nephropathy. Am J Physiol 1999;277:F766–F772.
[Article] [PubMed]58. Lan HY, Nikolic-Paterson DJ, Zarama M, Vannice JL, Atkins RC. Suppression of experimental crescentic glomerulonephritis by the interleukin-1 receptor antagonist. Kidney Int 1993;43:479–485.
[Article] [PubMed]59. Liu Y. Hepatocyte growth factor and the kidney. Curr Opin Nephrol Hypertens 2002;11:23–30.
[Article] [PubMed]60. Liu Y. Hepatocyte growth factor in kidney fibrosis: therapeutic potential and mechanisms of action. Am J Physiol Renal Physiol 2004;287:F7–F16.
[Article] [PubMed]61. Yang J, Liu Y. Delayed administration of hepatocyte growth factor reduces renal fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol 2003;284:F349–F357.
[Article] [PubMed]62. Mizuno S, Nakamura T. Suppressions of chronic glomerular injuries and TGF-beta 1 production by HGF in attenuation of murine diabetic nephropathy. Am J Physiol Renal Physiol 2004;286:F134–F143.
[Article] [PubMed]63. Li Y, Yang J, Dai C, Wu C, Liu Y. Role for integrin-linked kinase in mediating tubular epithelial to mesenchymal transition and renal interstitial fibrogenesis. J Clin Invest 2003;112:503–516.
[Article] [PubMed] [PMC]64. Liu Y, Sun AM, Dworkin LD. Hepatocyte growth factor protects renal epithelial cells from apoptotic cell death. Biochem Biophys Res Commun 1998;246:821–826.
[Article] [PubMed]65. Gong R, Rifai A, Tolbert EM, Centracchio JN, Dworkin LD. Hepatocyte growth factor modulates matrix metalloproteinases and plasminogen activator/plasmin proteolytic pathways in progressive renal interstitial fibrosis. J Am Soc Nephrol 2003;14:3047–3060.
[Article] [PubMed]66. Motazed R, Colville-Nash P, Kwan JT, Dockrell ME. BMP-7 and proximal tubule epithelial cells: activation of multiple signaling pathways reveals a novel anti-fibrotic mechanism. Pharm Res 2008;25:2440–2446.
[Article] [PubMed]67. Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, et al. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med 2003;9:964–968.
[Article] [PubMed]68. Wang S, Hirschberg R. BMP7 antagonizes TGF-beta-dependent fibrogenesis in mesangial cells. Am J Physiol Renal Physiol 2003;284:F1006–F1013.
[Article] [PubMed]69. Chevalier RL, Goyal S, Kim A, Chang AY, Landau D, LeRoith D. Renal tubulointerstitial injury from ureteral obstruction in the neonatal rat is attenuated by IGF-1. Kidney Int 2000;57:882–890.
[Article] [PubMed]71. Edgtton KL, Gow RM, Kelly DJ, Carmeliet P, Kitching AR. Plasmin is not protective in experimental renal interstitial fibrosis. Kidney Int 2004;66:68–76.
[Article] [PubMed]
Fig. 1
Pathologic findings of contralateral (A) and ipsilateral (B) kidney in C57BL/6 mice with UUO (day 7). Contralateral kidney without UUO shows intact glomerular and tubulointerstitial structure (A), whereas ipsilateral kidney with UUO shows tubulointerstitial fibrosis, tubular atrophy, and glomerulosclerosis (B). Kidney sections (2-µm thickness) were stained with Masson trichrome staining. Abbreviation: UUO, unilateral ureteral obstruction.
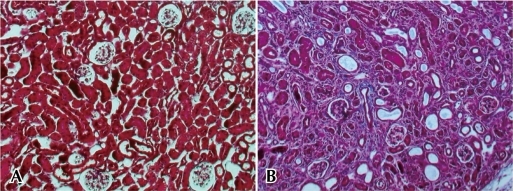
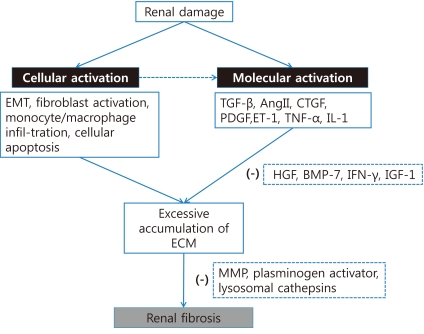
Fig. 2
Pathogenetic mechanism underlying renal fibrosis. Initial renal damage can induce both cellular and molecular activation. Cellular activation may subsequently provoke the activation of signal molecules. Both cellular activations such as EMT, fibroblast activation, monocyte/macrophage infiltration, and cellular apoptosis and molecular activations such as TGF-β, AngII, CTGF, PDGF, ET-1, TNF-α, and IL-1 result in the excessive accumulation of ECM. Antifibrotic cytokines such as HGF, BMP-7, IFN-γ, and IGF-1 or several proteases such as MMP, plasminogen activator, and lysosomal cathepsins may attenuate renal fibrosis. Modified from Eddy SS15). Abbreviations: EMT: epithelial-to-mesenchymal transition, TGF-β: transforming growth factor beta, AngII: angiotensin II, CTGF: connective tissue growth factor, PDGF: platelet-derived growth factor, ET-1: endothelin-1, TNF-α: tumor necrosis factor alpha, IL-1: interleukin 1, ECM: extracellular matrix, HGF: hepatocyte growth factor, BMP-7: bone morphogenetic protein 7, IFN-γ: interferon gamma, IGF-1: insulin-like growth factor 1, and MMP: matrix metalloproteinases.