About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 53(7); 2010
Effect of p16 on glucocorticoid response in a B-cell lymphoblast cell line
- Corresponding author: Kyung Yil Lee, M.D. Department of Pediatrics, The Catholic University of Korea, Daejeon St. Mary's Hospital, 520-2 Daeheung-dong, Jung-gu, Daejeon 301-723, Korea. Tel: +82.42-220-9541, Fax: +82.42-221-2925, leekyungyil@catholic.ac.kr
- Received February 02, 2010 Revised April 19, 2010 Accepted May 18, 2010
- Abstract
-
- Purpose
- Purpose
- It has been suggested that p16 has a role in glucocorticoid (GC)-related apoptosis in leukemic cells, but the exact mechanisms have yet to be clarified. We evaluated the relationship between the GC response and p16 expression in a lymphoma cell line.
- Methods
- Methods
- We used p16 siRNA transfection to construct p16-inactivated cells by using the B-cell lymphoblast cell line NC-37. We compared glucocorticoid receptor (GR) expression, apoptosis, and cell viability between control (p16+ NC-37) and p16 siRNA-transfected (p16- NC-37) cells after a single dose of dexamethasone (DX).
- Results
- Results
- In both groups, there was a significant increase in cytoplasmic GR expression, which tended to be higher for p16+ NC-37 cells than for p16- NC37 cells at all times, and the difference at 18 h was significant (P<0.05). Similar patterns of early apoptosis were observed in both groups, and late apoptosis occurred at higher levels at 18 h when the GR had already been downregulated (P<0.05). Cell viability decreased in both groups but the degree of reduction was more severe in p16+ NC-37 cells after 18 h (P<0.05).
- Conclusion
- Conclusion
- These results suggest a relationship between GR expression and cell cycle inhibition, in which the absence of p16 leads to reduced cell sensitivity to DX.
- Introdction
- Introdction
Glucocorticoid (GC) is an important chemotherapeutic agent used to treat leukemias and lymphomas1-3). GC combines with a cytoplasmic glucocorticoid receptor (GR) that binds to specific DNA sequences as a transcription factor. The liganded receptors also bind to and interfere with other transcription factors, such as activator protein-1 and nuclear factor-[kappa]B, and inhibit the mitogen-activated protein kinase pathways that mediate the expression of many of the genes involved in inflammatory and immune responses, including apoptosis and cell cycle arrest4, 5). Clinically, GC sensitivity is an important factor determining prognosis of acute lymphoblastic leukemia (ALL), and non-responsiveness to GC is used as a marker for classifying patients into risk groups1, 6). However, the molecular mechanisms of the anti-leukemic effects and the clinically important phenomenon of GC resistance require further study4, 5, 7).Progression from the G1 to S phase of the cell cycle is regulated by a series of structurally related enzymes: cyclin regulates the activation of cyclin-dependent kinases (CDKs); CDKs regulate the retinoblastoma protein (pRb) and induce the subsequent release of E2F transcription factors and expression of the genes required for the S phase. Cyclin-CDK complexes are regulated negatively by a family of kinase inhibitors8, 9). The p16INK4 (CDKN2A) gene located on chromosome 9p21 encodes a protein (p16) that inhibits CDKs and that can block cell cycle progression8-10). Frequently, p16 is mutated or inactivated in primary tumors, including leukemias, lymphomas, gliomas, lung carcinomas, and many cancer cell lines; it is thus regarded as a tumor suppressor gene11-13). Recently, some studies have reported that p16 is associated with the prognosis of hematologic malignancies, although this is controversial11). It has also been suggested that p16 has a role in GC-related apoptosis in leukemic cells, and the inactivation of p16 in B-cell ALL may induce cells that are more resistant to GC14, 15). However, few studies have examined the relationship between GC responsiveness and p16, and the roles and exact mechanisms of p16 in hematologic malignancies are not clear.This study evaluated the relationship between GC responses, including GR expression and subsequent apoptosis, and p16, using the B-cell lymphoblast cell line NC-37.
- Materials and methods
- Materials and methods
- 1. Cell line, culture conditions, and reagents
- 1. Cell line, culture conditions, and reagents
The B-cell lymphoblast cell line NC-37 (ATCC number, CCL-214) was purchased from ATCC (Rockville, MD, USA). NC-37 cells were maintained in RPMI-1640 medium (Gibco BRL Life Technologies, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Gibco BRL, Rockville, MD, USA) at 5% CO2 and 37℃ at saturated humidity.- 2. p16 siRNA transfection
- 2. p16 siRNA transfection
For p16 siRNA transfection, a commercial kit was used (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Briefly, the following solutions were prepared: solution A contained 7 µL p16 siRNA (sc-36143) or control siRNA (sc-36869 or sc-37007) in 100 µL siRNA transfection medium (sc-36868). Solution B contained 6 µL siRNA transfection reagent (sc-29528) in 100 µL siRNA transfection medium.Solution A was added to solution B directly, and the mixture was incubated for 30 min at room temperature. For each transfection, 0.8 mL siRNA transfection medium was added to each tube containing the siRNA and transfection reagent mixtures. In 6-well tissue plates, 3×105 cells were seeded per well and the mixtures were overlaid onto the cells. The cells were incubated for 6 h at 37℃ in a CO2 incubator, and then the transfection mixtures were removed and replaced with RPMI-1640 medium supplemented with 10% FBS and then incubated for an additional 6 h.- 3. Western blot analysis
- 3. Western blot analysis
Western blotting was used to detect p16 protein after p16 siRNA transfection. Experiments were done for wild-type, control, and p16 siRNA-transfected NC-37 cells. The cells were collected by centrifugation, washed in phosphate-buffered saline (PBS), and lysed by the addition of SDS sample buffer (62.5 mM Tris-HCl [pH 6.8], 6% [w/v] SDS, 30% glycerol, 125 mM DTT, and 0.03% [w/v] bromophenol blue). Total cell samples were lysed and denatured by boiling for 5 min at 100℃. Equal amounts of protein from each sample were separated by 15% SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (Bio-Rad Laboratories, Hercules, CA, USA). The membranes were blocked for 1 h with Tris-buffered saline containing 5% (w/v) milk and 0.1% Tween 20, then incubated with the primary rabbit monoclonal antibody for p16 (p16 INK4A Antibody; Cell Signaling Technology, Beverly, MA, USA) overnight at 4℃. The blots were washed with Tris-buffered saline containing Tween 20, incubated with the anti-rabbit secondary antibody (Cell Signaling Technology, Beverly, MA, USA) for 2 h, and developed using West-Zol TM plus (iNtRON Biotechnology, Seoul, Korea).- 4. Cell culture with dexamethasone (DX)
- 4. Cell culture with dexamethasone (DX)
DX purchased from Sigma Chemical (St. Louis, MO, USA) was dissolved in dimethyl sulfoxide and added to the medium after p16 siRNA transfection. To study the effect of DX on p16 status, we added DX to samples containing wild-type, control, and p16 siRNA-transfected NC-37 cells. The final concentration of DX was adjusted to 100 nM. For measurements, cells were harvested 6, 12, 18, and 24 h after DX addition and then prepared for the next steps.- 5. Flow cytometry analysis of GR expression
- 5. Flow cytometry analysis of GR expression
Cultured cells were washed twice in PBS containing 1% bovine serum albumin. Aliquots of 1×106 cells were fixed in paraformaldehyde at room temperature for 30 min, washed, and permeabil ized with 0.1% Triton X-100 in 0.1% citrate buffer for 5 min on ice. The cells were washed twice and incubated at 4℃ for 70 min with either FITC-conjugated anti-GR antibody (5E4; AbD Serotec, Oxford, UK) or FITC-conjugated isotype control (IgG1 AbD Serotec, Oxford, UK). Finally, the cells were washed and resuspended in PBS containing 1% bovine serum albumin and analyzed using a Coulter Elite flow cytometer (Beckman Coulter Inc., Fullerton, CA, USA).- 6. Cell apoptosis test
- 6. Cell apoptosis test
To detect apoptosis, we performed double staining with FITC-Annexin V and propidium iodide (PI; R & D Systems, Minneapolis, MN, USA). First 1×106 cells were washed with PBS. FITC-Annexin V was diluted at a concentration of 1 mg/mL in binding buffer, and the cells were resuspended in 1 mL of this solution (prepared fresh each time). The resuspended cells were incubated for 10 min in the dark at room temperature, and then 0.1 mL PI solution was added to the cell suspensions before analysis to give a final concentration of 1 mg/mL. These cells were analyzed on using a Coulter Elite flow cytometer. Annexin V single-positive cells were regarded as early apoptotic cells, whereas Annexin V/PI double-positive cells were regarded as late apoptotic cells.- 7. Alamar blue (AB) assay for cell viability
- 7. Alamar blue (AB) assay for cell viability
First 1×104 cells were suspended in 95 µL phenol red-free RPMI-1640 containing 0.1% FBS, then seeded into 96-well plates. DX was added to each plate at a concentration of 100 nM. After 6, 12, 18, and 24 h, 11 µL AB solution was added to the medium directly, resulting in a final concentration of 10%. As a negative control, AB was added to medium without cells. Then the plates were incubated for 4 h at 37℃. The absorbance of test and control wells was read at 570 and 595 nm with a standard spectrophotometer. The number of viable cells correlated with the magnitude of dye reduction and is expressed as a percentage of the AB reduction compared to control AB.- 8. Statistics
- 8. Statistics
All statistical analyses were conducted using SPSS 13.0 (SPSS, Chicago, IL, USA). The results were expressed as the mean±standard deviation, and the differences between the cell groups at each time point were analyzed using the Mann-Whitney U-test. General repeated measured analysis of variance was applied to identify the trends of time-dependent GR expression, apoptosis, and cell viability between the two groups. P value <0.05 was regarded as statistically significant.
- Results
- Results
- 1. Identification of p16 RNA interference in NC-37
- 1. Identification of p16 RNA interference in NC-37
To determine whether p16 affects GR regulation and the apoptosis of lymphoblast cells, we generated p16 siRNA-transfected NC-37 cells that did not express p16. First we used the florescence-expressing control siRNA to confirm p16 siRNA transfection. When more than 50% of the control siRNA-transfected NC-37 cells had fluoresced, Western blot analysis was performed to detect p16. The wild-type and control NC-37 cells expressed p16 in the immunoblot analysis, whereas the p16 siRNA-transfected NC-37 did not (P<0.05, Fig. 1).- 2. GR expression after DX treatment
- 2. GR expression after DX treatment
We evaluated time-dependent GR expression 0, 6, 12, 18, and 24 h after treatment with DX for control (p16+ NC-37) and p16 siRNA-transfected (p16- NC-37) cells. The GR levels were determined by flow cytometer after intracellular GR staining at each time point. After DX treatment, GR expression began to increase after 6 h, reached a peak at 18 h, and decreased sharply by 24 h (P<0.05). The GR expression levels at 18 and 24 h in both groups differed statistically (P<0.05). The degree of GR expression tended to be higher for p16+ NC-37 cells than p16- NC-37 cells at all times, and the difference was significant at 18 h (P<0.05, Fig. 2).- 3. Apoptotic changes after DX treatment
- 3. Apoptotic changes after DX treatment
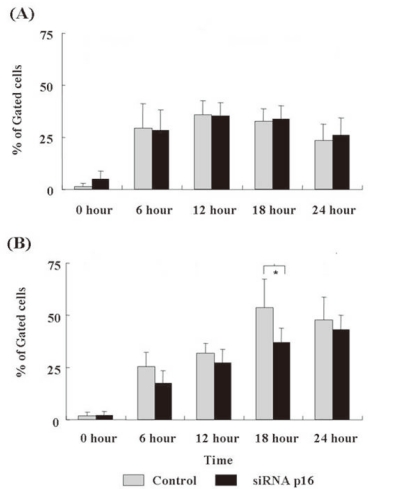
We assessed the time-dependent apoptotic changes after DX treatment using Annexin V/PI staining of cells by flow cytometry. After the DX treatment, both p16+ and p16- NC-37 cells showed a marked initial increase in Annexin V-stained cells (the early apoptotic cells) at 6 h, followed by a decrease at 24 h (both P<0.05). However, there was no statistical difference between the groups.The late apoptotic cells (double-positive cells) in p16- NC-37 cells increased through 6~18 h and reached a maximum at 18 h, and >50% of the cells were apoptotic. In contrast, the late apoptotic cells increased in a time-dependent manner over 24 h in p16- NC-37 cells. Overall, p16+ NC-37 cells was more susceptible to DX-induced late apoptosis than p16- NC-37 cells, and the result at 18 h was significant (P<0.05, Fig. 3).- 4. Time-dependent cell viability after DX treatment
- 4. Time-dependent cell viability after DX treatment
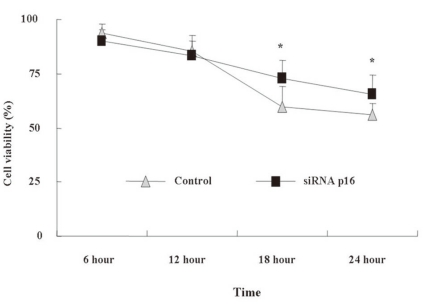
We assessed time-dependent cell viability using the AB assay. AB is a redox indicator that produces a colorimetric change in the fluorescent signal in response to metabolic activity. Within 12 h, the cell viability between p16+ and p16- NC-37 cells was not significantly different. At 18 and 24 h, the cell viability was reduced compared to the value at 12 h in both groups (P<0.05), and the degree of the decrease was more severe for p16+ NC-37 (P<0.05, Fig. 4).
- Discussion
- Discussion
Clinically, GC resistance indicates a poor prognosis in ALL treatment1-3). However, the molecular mechanisms of the anti-leukemic effects and GC resistance are not clear3-5). It has been hypothesized that GC induces apoptosis by affecting the Bcl-2 gene directly or inducing the release of apoptogenic factors. In addition, GC-induced cell cycle arrest in leukemic cells may be independent of apoptosis induction and associated with increased expression of CDK inhibitors such as p273-5). Factors affecting GC sensitivity include the availability of the hormone, tissue-specific factors, the intracellular metabolism of the hormones, and GR responses7). There is increasing evidence that the level of GR expression auto-induced by GC is associated with GR molecular functions, including steroid responsiveness and resistance5, 14, 15).The inactivation of p16 has been confirmed to varying degrees in T-cell leukemias and other hematologic malignancies16-23), and INK4A gene knockout mice develop spontaneous lymphomas at high frequency24). These findings suggest that p16 is involved in the negative regulation of tumor proliferation and progression in the lymphoid lineage. Recently, p16 was also studied as a prognostic factor for the diagnosis of hematologic malignancies3, 13). It was reported that the inactivation of p16 might be associated with the presence of minimal residual disease at the induction of chemotherapy, and loss of p16 function might also be a marker of chemoresistance in non-high-risk B-cell ALL3). However, other studies of several hematologic malignancies do not confirm these findings11). Ausserlechner et al reported that p16 sensitizes lymphoblastic leukemia cells to apoptosis by GC14). Using p16 gene transfection to GC-sensitive T lymphoma cell lines, they observed that after GC treatment the p16+ cells showed increased GR expression and sensitivity to GC, whereas the p16- cells did not. They postulated that because leukemic cells may have a program for escaping apoptosis by suppressing apoptotic pathways, p16 inactivation might be required for lymphoid malignancies to escape from as yet unrecognized tumor surveillance, including GC-induced apoptosis14). However, the mechanisms of p16 inactivation contributing to GR regulation and subsequent apoptosis remain unknown.Few studies have examined the relationship between GC responsiveness and p16. In this study, we evaluated the time course of GC-induced GR expression changes and the effect of p16 on GC-induced apoptosis using p16 siRNA transfection of a B-cell lymphoblast cell line, NC-37 cells. We found a pattern of intracytoplasmic GR expression after DX treatment within 24 h. That is, the initial GR levels peaked at 18 h, followed by a sudden decrease at 24 h in p16+ and p16- NC-37 cells; the former tended to show higher expression rates (Fig. 2). The repression of GR expression 24 h after GC treatment has also been observed in other studies25-27). Along with GR expression, similar patterns of early apoptotic cells induced by DX were observed in both groups (Fig. 3A). However, late apoptotic cells increased in a time-dependent manner, and this was more marked at 18 h for p16+ NC-37 cells. This suggests that p16 influences DX-induced late apoptosis more than early apoptosis (Fig. 3B). The viable cell assay showed similar results for both groups (Fig. 4).Combined, our results suggest that p16 is positively correlated with GR expression and GC-induced late apoptosis. Further effort is necessary to identify additional genetic changes in cell cycle regulators to complement these findings. Studies of p16 and GC responsiveness may lead to new treatment modalities, such as a combination of GC with substances mimicking p16 function, including CDK4- or CDK6-inhibiting peptides, for hematologic malignancies that do not express p1614). In addition, because GR expression is not the sole factor determining GC sensitivity4-6), further studies must examine the relationship between GC-induced apoptosis and the interaction of other signaling events associated with p16 or other proteins.In conclusion, although p16 has a role in GR expression and apoptosis induced by GC, it remains to be seen whether the GC-induced apoptosis mechanism can interact with the other signaling events associated with the p16 gene operating during such malignant changes. This observation might have important implications for cancer therapy.
- References
- 1. Biondi A, Valsecchi MG, Seriu T, D'Aniello E, Willemse MJ, Fasching K, et al. Molecular detection of minimal residual disease is a strong predictive factor of relapse in childhood B-lineage acute lymphoblastic leukemia with medium risk features. A case control study of the International BFM Study Group. Leukemia 2000;14:1939–1943.
[Article] [PubMed]2. Myoumoto A, Nakatani K, Koshimizu TA, Matsubara H, Adachi S, Tsujimoto G. Glucocorticoid-induced granzyme A expression can be used as a marker of glucocorticoid sensitivity for acute lymphoblastic leukemia therapy. J Hum Genet 2007;52:328–333.
[Article] [PubMed]3. Tutor O, Díaz MA, Ramírez M, Algara P, Madero L, Martínez P. Loss of heterozygosity of p16 correlates with minimal residual disease at the end of the induction therapy in non-high risk childhood B-cell precursor acute lymphoblastic leukemia. Leuk Res 2002;26:817–820.
[Article] [PubMed]4. Frankfurt O, Rosen ST. Mechanisms of glucocorticoid-induced apoptosis in hematologic malignancies: updates. Curr Opin Oncol 2004;16:553–563.
[Article] [PubMed]5. Renner K, Ausserlechner MJ, Kofler R. A conceptual view on glucocorticoid-induced apoptosis, cell cycle arrest and glucocorticoid resistance in lymphoblastic leukemia. Curr Mol Med 2003;3:707–717.
[Article] [PubMed]6. Jenkins BD, Pullen CB, Darimont BD. Novel glucocorticoid receptor coactivator effector mechanisms. Trends Endocrinol Metab 2001;12:122–126.
[Article] [PubMed]7. Bladh LG, Lidén J, Dahlman-Wright K, Reimers M, Nilsson S, Okret S. Identification of endogenous glucocorticoid repressed genes differentially regulated by a glucocorticoid receptor mutant able to separate between nuclear factor-kappaB and activator protein-1 repression. Mol Pharmacol 2005;67:815–826.
[Article] [PubMed]8. Guo SX, Taki T, Ohnishi H, Piao HY, Tabuchi K, Bessho F, et al. Hypermethylation of p16 and p15 genes and RB protein expression in acute leukemia. Leuk Res 2000;24:39–46.
[Article] [PubMed]9. Chim CS, Wong AS, Kwong YL. Epigenetic inactivation of the CIP/KIP cell-cycle control pathway in acute leukemias. Am J Hematol 2005;80:282–287.
[Article] [PubMed]10. Chim CS, Wong KY, Loong F, Lam WW, Srivastava G. Frequent epigenetic inactivation of Rb1 in addition to p15 and p16 in mantle cell and follicular lymphoma. Hum Pathol 2007;38:1849–1857.
[Article] [PubMed]11. Drexler HG. Review of alterations of the cyclin-dependent kinase inhibitor INK4 family genes p15, p16, p18 and p19 in human leukemia-lymphoma cells. Leukemia 1998;12:845–859.
[Article] [PubMed]12. Suga Y, Miyajima K, Oikawa T, Maeda J, Usuda J, Kajiwara N, et al. Quantitative p16 and ESR1 methylation in the peripheral blood of patients with non-small cell lung cancer. Oncol Rep 2008;20:1137–1142.
[PubMed]13. de Snoo FA, Bishop DT, Bergman W, van Leeuwen I, van der Drift C, van Nieuwpoort FA, et al. Increased risk of cancer other than melanoma in CDKN2A founder mutation (p16-Leiden)-positive melanoma families. Clin Cancer Res 2008;14:7151–7157.
[Article] [PubMed]14. Ausserlechner MJ, Obexer P, Wiegers GJ, Hartmann BL, Geley S, Kofler R. The cell cycle inhibitor p16 (INK4A) sensitizes lymphoblastic leukemia cells to apoptosis by physiologic glucocorticoid levels. J Biol Chem 2001;276:10984–10989.
[Article]15. Holleman A, den Boer ML, Kazemier KM, Janka-Schaub GE, Pieters R. Resistance to different classes of drugs is associated with impaired apoptosis in childhood acute lymphoblastic leukemia. Blood 2003;102:4541–4546.
[Article] [PubMed]16. Ogawa S, Hirano N, Sato N, Takahashi T, Hangaishi A, Tanaka K, et al. Homozygous loss of the cyclin-dependent kinase 4-inhibitor (p16) gene in human leukemias. Blood 1994;84:2431–2435.
[Article] [PubMed]17. Cayuela JM, Madani A, Sanhes L, Stern MH, Sigaux F. Multiple tumor-suppressor gene 1 inactivation is the most frequent genetic alteration in T-cell acute lymphoblastic leukemia. Blood 1996;87:2180–2186.
[Article] [PubMed]18. Ohnishi H, Kawamura M, Ida K, Sheng XM, Hanada R, Nobori T, et al. Homozygous deletions of p16/MTS1 gene are frequent but mutations are infrequent in childhood T-cell acute lymphoblastic leukemia. Blood 1995;86:1269–1275.
[Article] [PubMed]19. Gombart AF, Morosetti R, Miller CW, Said JW, Koeffler HP. Deletions of the cyclin-dependent kinase inhibitor genes p16INK4A and p15INK4B in non-Hodgkin's lymphomas. Blood 1995;86:1534–1539.
[Article] [PubMed]20. Stranks G, Height SE, Mitchell P, Jadayel D, Yuille MA, De Lord C, et al. Deletions and rearrangement of CDKN2 in lymphoid malignancy. Blood 1995;85:893–901.
[Article] [PubMed]21. Uchida T, Watanabe T, Kinoshita T, Murate T, Saito H, Hotta T. Mutational analysis of the CDKN2 (MTS1/p16ink4A) gene in primary B-cell lymphomas. Blood 1995;86:2724–2731.
[Article] [PubMed]22. Koduru PR, Zariwala M, Soni M, Gong JZ, Xiong Y, Broome JD. Deletion of cyclin-dependent kinase 4 inhibitor genes P15 and P16 in non-Hodgkin's lymphoma. Blood 1995;86:2900–2905.
[Article] [PubMed]23. Dreyling MH, Bohlander SK, Le Beau MM, Olopade OI. Refined mapping of genomic rearrangements involving the short arm of chromosome 9 in acute lymphoblastic leukemias and other hematologic malignancies. Blood 1995;86:1931–1938.
[Article] [PubMed]24. Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell 1996;85:27–37.
[Article] [PubMed]25. Okret S, Poellinger L, Dong Y, Gustafsson JA. Down-regulation of glucocorticoid receptor mRNA by glucocorticoid hormones and recognition by the receptor of a specific binding sequence within a receptor cDNA clone. Proc Natl Acad Sci USA 1986;83:5899–5903.
[Article] [PubMed] [PMC]26. Rosewicz S, McDonald AR, Maddux BA, Goldfine ID, Miesfeld RL, Logsdon CD. Mechanism of glucocorticoid receptor down-regulation by glucocorticoids. J Biol Chem 1988;263:2581–2584.
[Article] [PubMed]27. Hoeck W, Rusconi S, Groner B. Down-regulation and phosphorylation of glucocorticoid receptors in cultured cells. Investigations with a monospecific antiserum against a bacterially expressed receptor fragment. J Biol Chem 1989;264:14396–14402.
[PubMed]
Fig. 1
Western blot analysis of p16 compared with β-actin in NC-37 cells. Wild-type, control siRNA-transfected, and p16 siRNA-transfected NC-37 cells were immunoblotted with p16 antibody. The p16 siRNA-transfected NC-37 cells did not express p16 protein. The bar graphs express the ratio of p16 to β-actin calculated from densitometry measurements. *P<0.05.

Fig. 2
Cytoplasmic glucocorticoid receptor (GR) expression levels as measured by flow cytometry. Time-dependent changes in GR expression after dexamethasone (DX) treatment are shown for control and p16 siRNA-transfected NC-37 (A) along with the results of flow cytometry (B). GR expression peaked at 18 h and decreased sharply at 24 h. The control NC-37 cells expressed higher glucocorticoid receptor (GR) levels compared to the p16 siRNA-transfected NC-37 cells at 18 h. *P<0.05.
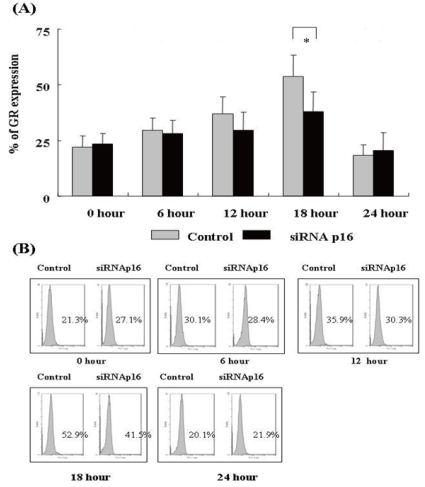
Fig. 3
Apoptotic cells stained with annexin V and propidium iodide (PI) as assessed by flow cytometry in both groups. Annexin V single-positive cells were regarded as early apoptotic cells (A) and double-positive cells were regarded as late apoptotic cells (B). There were no statistical differences between the 2 groups for early apoptosis (A). Late apoptotic cells increased in a time-dependent manner and peaked at 18 h in the control NC-37 cells. *P<0.05.