About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 53(7); 2010
A case of regression of atypical dense deposit disease without C3 deposition in a child
- Corresponding author: Dae-Yeol Lee, M.D, Ph.D. Department of Pediatrics, Chonbuk National University Hospital, 634-18, Keumam-dong, Jeonju 561-712, Korea. Tel: +82.63-250-1469, Fax: +82.63-250-1464, leedy@chonbuk.ac.kr
- Received March 11, 2010 Revised April 13, 2010 Accepted May 18, 2010
- Abstract
-
Dense deposit disease (DDD) is a rare disorder characterized by the deposition of abnormal electron-dense material within the glomerular basement membrane of the kidneys. The diagnosis is made in most patients between 5 and 15 years of age, and within 10 years, approximately half of the affected patients progress to end-stage renal disease. We report a rare case of regressive DDD without C3 deposition after steroid therapy in an 11-year-old boy. The patient presented with edema, gross hematuria, and nephrotic-range proteinuria. Laboratory testing revealed a serum creatinine level of 1.17 mg/dL, albumin level of 2.3 g/dL, and serum C3 level of 125 mg/dL (range 90-180 mg/dL). The results of the renal biopsy were consistent with DDD without C3 deposition. After 6 weeks of steroid therapy, the nephrotic syndrome completely resolved. The follow-up renal biopsy showed a significant reduction in mesangial proliferation and disappearance of electron-dense deposits in the GBM.
- Introdction
- Introdction
Membranoproliferative glomerulonephritis (MPGN) is a chronic progressive renal disease that is diagnosed by renal histological features. On the basis of immunopathology and ultrastructure analysis of the kidney and of the glomerulus, three subtypes are recognized. MPGN types I and III are variants of immune complex-mediated disease; MPGN II, which is also known as dense deposit disease (DDD), by contrast, has no known association with immune complexes. Because recent studies have indicated that the light microscopy pattern in most patients is not membranoproliferative, the recent trend is to consider DDD a distinct entity, rather than a variant of MPGN1). MPGN II is rare, and it accounts for less than 20% of all cases of MPGN in children2). Its morphologic hallmark is the presence of dense deposits within the glomerular basement membrane (GBM) as observed by electron microscopy.MPGN II has a poor prognosis and, with few exceptions, there is usually no change in the dense deposits on follow-up2, 3). Although there is no universally effective treatment for MPGN II, some reports4, 5) suggest that prednisone therapy can reduce proteinuria and prolong renal survival.We report the case of a child with atypical DDD who achieved complete remission, clinical and morphological, after steroid therapy.
- Case report
- Case report
An 11-year-old boy presented with gross hematuria, proteinuria and generalized edema for five days. There had been no systemic illness, rash or joint pain. His past and family histories were not remarkable for renal disease. Laboratory data included a white blood cell count of 7,000/mm3 with a normal differential count, hemoglobin 11.0 g/dL, and a platelet count of 337,000/mm3. The total serum protein was 4.7 g/dL, with a serum albumin concentration of 2.3 g/dL. The serum creatinine level was 1.17 mg/dL, cholesterol 525 mg/dL and triglycerides 153 mg/dL. The glomerular filteration rate was 66.3 ml/min/1.73 m2. Urinalysis showed hematuria and proteinuria (3+), and the 24 hour urinary protein excretion was 2,625 mg. The serum C3 level was 125 mg/dL (range 90-180 mg/dL) and the C4 level was 33.6 mg/dL (range 10-40 mg/dL). The anti-nuclear antibodies, anti-neutrophil cytoplasm antibodies, anti-hepatitis B virus, Epstein-Barr virus, cytomegalovirus antibodies, and anti-streptolysin O titer were all negative.The patient underwent a renal biopsy on the fourth hospital day. On light microscopy, the biopsy showed 43 glomeruli. Some glomeruli had mesangial hypercellularity or lobular accentuation (Fig. 1). The glomerular capillary walls were thickened and showed a segmental double contour. In addition, mild interstitial fibrosis and tubular atrophy were present. The ultrastructural examination showed wide foot process effacement, irregularly thickened glomerular basement membrane (GBM), and electron-dense deposits in the lamina densa (Fig. 2). Immunofluorescence examination was negative for immunoglobulin (Ig) G, Ig A, Ig M and C3 deposition. Even though the serum C3 level was not low and there was no C3 deposition along the capillary walls, the pathology was consistent with DDD.Daily prednisolone therapy (60 mg/m2/day) was started with dipyridamole. After 28 days of daily prednisone therapy, the prednisolone was reduced to alternate day treatment (40 mg/m2/day) and then gradually weaned. Six weeks after steroid therapy, the nephrotic syndrome was in remission with no further relapse. However, he continued to have microscopic hematuria for 16 months after disease onset. At 18 months after onset of the glomerulonephritis, a follow-up renal biopsy was performed. The second renal biopsy showed significant reduction of mesangial proliferation with mild mesangial expansion and disappearance of the electron-dense deposits in the GBM (Fig. 3). In contrast to the first biopsy, mild Ig M staining (1+) was detected in the mesangium. Currently, the patient has been monitored for seven years; he remains well, and has no urinary abnormalities.
- Discussion
- Discussion
This is a very rare case of atypical DDD that showed complete resolution of the dense deposits after steroid therapy. The natural history of idiopathic DDD was recorded in this 11 year old boy that followed for seven years and showed complete resolution of DDD after high dose prednisolone therapy. The first reported case of MPGN II was described in 19626). However, the natural history of MPGN II remains unclear.Clinical manifestations at diagnosis, such as the nephrotic syndrome, hypertension, persistent hypocomplementemia and the biopsy findings of glomerular crescents or tubular atrophy have been previously cited as indicators of a poor outcome2, 3). Recently, Nasr et al7) reported that an older age and higher serum creatinine at biopsy are predictors of a poor prognosis in patients with DDD. Taking into account the prior reports of predictive factors, our patient had a good prognosis because of the young age, normal blood pressure, normal C3 level, and absence of glomerular crescents. In addition, absence of C3 deposition on GBM may be a predictable marker for good prognosis as in our case.Distinct features of MPGN II include the complement profile and the frequent presence of the nephritic factor (NF). The profile usually includes C3 levels that are markedly depressed with normal or near normal levels of other components8). In more than 50% of patients, the serum C3NF persists throughout the disease course9). C3NF is always associated with clinical evidence of complement activation such as a reduction in CH50 and a decrease in C3. However, the relationship among C3NF, C3 levels and prognosis is unclear. Some groups report no correlation between C3 levels and clinical course7, 10), whereas other groups have found persistent hypocomplementemia indicative of a poor prognosis3, 11). Klein M et al.12) showed that the DDD patients with normal amounts of C3 and factor B, and low levels of C3NF activity had a benign course. The level of C3 in our patient was not decreased throughout the course of the disease over the seven years of follow up, but factor B and C3NF were not checked.The dense intramembranous deposits in the GBM are the pathognomonic feature of MPGN II, and are distributed in a segmental, discontinuous, or diffuse pattern in the lamina densa of the GBM. The GBM is not a static structure, and the dense deposits represent a biochemically altered BM substance13). Tornroth and Skrifvars14) suggested a slow turnover of GBM with removal of deposit material on the endothelial side of the GBM in patients with gold-induced membranous nephropathy. Spontaneous remissions of MPGN II are uncommon, and electron dense deposits usually are still present on follow-up renal biopsies. To date, spontaneous improvement, clinical and morphological, has been reported in only a few cases of MPGN II in children4, 15). McEnery and McAdams4) reported that six children with MPGN II showed significant reduction in pathological changes, and complete resolution of the dense deposits were observed in two patients that had normal C3 levels. The improvement of the clinical status with steroid therapy, normal serum C3 level and no C3 deposition on GBM, in our patient, might predict the possibility of changes in glomerular morphology including resolution of the dense deposits on follow-renal biopsy. In this patient, significant reduction of mesangial proliferation and complete regression of the dense deposits was observed 18 months after disease onset. The present observations suggest that the renal pathology associated with DDD is fundamentally transient and that resolution, if it occurs, does so slowly over several years.Various treatment modalities, including immunosuppression with corticosteroids, cytotoxic drugs and cyclosporin A, anticoagulants and antiplatelet therapies are used for DDD with varying degrees of success3). The efficacy of steroid therapy is controversial. McEnery and McAdams4) reported improvement in glomerular morphology and a good clinical outcome with long-term alternate-day prednisone therapy. Our patient showed clinical remission within six weeks of steroid therapy. Whether clinical and morphological regression were due to steroid therapy is difficult to confirm. However, steroid therapy may promote clinical remission and spontaneous removal of dense deposit by modification or elimination of the nephritic elements associated with this disease.
- References
- 1. Walker PD. Dense deposit disease: New insights. Curr Opin Nephrol Hypertens 2007;16:204–212.
[Article] [PubMed]2. Habib R, Gubler M-C, Loirat C, Ben Maiz H, Levy M. Dense deposit disease: a variant of membranoproliferative glomerulonephritis. Kidney Int 1975;7:204–215.
[Article] [PubMed]3. Appel GB, Cook HT, Hageman G, Jennette C, Kashgarian M, Kirshfink M, et al. Membranoproliferative glomerulonephritis type II(dense deposit disease): An update. J Am Soc Nephrol 2005;16:1392–1404.
[Article] [PubMed]4. McEnery PT, McAdams AJ. Regression of membranoproliferative glomerulonephritis type II(dense deposit disease): observation in six children. Am J Kidney Dis 1988;12:138–146.
[Article] [PubMed]5. The Southwest Pediatric Nephrology Study Group. Dense deposit disease in children: prognostic value of clinical and pathologic indicators. Am J Kidney Dis 1985;6:161–169.
[Article] [PubMed]6. Berger J, Galle P. Alteration sunguliere des membranes basales du rein. J Urol Nephrol (Paris) 1962;68:116
[PubMed]7. Nasr SH, Valeri AM, Appel GB, Sherwinter J, Stokes MB, Said SM, et al. Dense deposit disease: clinicopathologic study of 32 pediatric and adult patients. Clin J Am Soc Nephrol 2009;4:22–32.
[Article] [PubMed] [PMC]8. Varade WS, Forristal J, West CD. Patterns of complement activation in idiopathic membranoproliferative glomerulonephritis, type I, II, and III. Am J Kidney Dis 1990;16:196–206.
[Article] [PubMed]9. Schwertz R, Rother U, Anders D, Gretz N, Scharer K, Kirschfink M. Complement analysis in children with idiopathic membranoproliferative glomerulonephritis: A long-term follow-up. Pediatr Allergy Immunol 2001;12:166–172.
[Article] [PubMed]10. Davis AE, Schneeberger EE, Grupe WW, McCluskey RT. Membranoproliferative glomerulonephritis(MPGN type I) and dense deposit disease(DDD) in children. Clin Nephrol 1978;9:184–193.
[PubMed]11. Kashtan CE, Burke B, Burch G, Gustav Fisker S, Kim Y. Dense intramembranous deposit disease: A clinical comparison of histological subtypes. Clin Nephrol 1990;33:1–6.
[PubMed]12. Klein M, Poucell S, Arbus GS, McGraw M, Rance CP, Yoon SJ, et al. Characteristics of a benign subtype of dense deposit disease: comparison with the progressive form of this disease. Clin Nephrol 1983;20:163–171.
[PubMed]13. Nevins TE. Lectin binding in membranoproliferative glomerulonephritis. Evidence for N-acetylglucosamine in dense intramembranous deposits. Am J Pathol 1985;118:325–330.
[PubMed] [PMC]14. Tornroth T, Skrifvaars B. The development and resolution of glomerular basement membrane changes associated with subepithelial immune deposits. Am J Pathol 1975;79:219–230.
[PubMed] [PMC]15. Kher KK, Makker SP, Aikawa M, Kirson IJ. Regression of dense deposits in type II membranoproliferative glomerulonephritis : case report of clinical course in a child. Clin Nephrol 1982;17:100–103.
[PubMed]
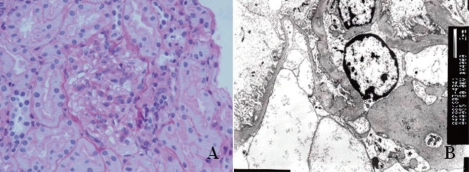
Fig. 1
Light microscopic features of dense deposit disease. A) Lobular configuration of the glomerular tuft due to expansion of the endocapillary zones by cells and matrix (periodic acid-Schiff stain, ×400) B) Mesangial hypercellularity accompanied by a mild increase in mesangial matrix (periodic acid-Schiff stain, ×400).
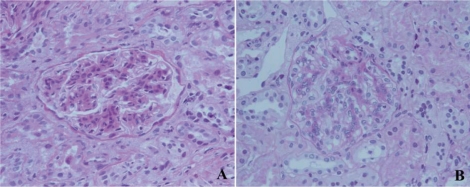
Fig. 2
Electron microscopic features of dense deposit disease: (A) Intramembranous electron-dense deposits thicken the glomerular basement membranes (×4000); (B) Discontinuous intramembranous rounded aggregates of electron-dense material (×4000).
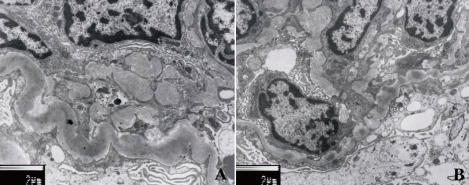
Fig. 3
Dense deposit disease after treatment. A) A second biopsy 18 months after treatment shows striking improvement in the severity of the glomerulonephritis with regression of the proliferation (periodic acid-Schiff stain, ×400). B) Electron micrograph shows focal foot process effacement, segmental irregular thickening of glomerular basement membrane, and no electron dense deposits (×4,000).