About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 53(7); 2010
A case of Pfeiffer syndrome with c833_834GC>TG (Cys278Leu) mutation in the FGFR2 gene
- Corresponding author: Jong Beom Sin, M.D. Department of Pediatrics, Busan Paik Hospital, Inje University College of Medicine, 633-165, Gaegeom 2dong, Busanjin gu, Busan 614-735, Korea. Tel: +82.51-890-6126, Fax: +82.51-895-7785, pedsin@inje.ac.kr
- Received February 22, 2010 Revised April 12, 2010 Accepted May 18, 2010
- Abstract
-
Pfeiffer syndrome is a rare autosomal dominant disorder characterized by coronal craniosynostosis, brachycephaly, mid-facial hypoplasia, and broad and deviated thumbs and great toes. Pfeiffer syndrome occurs in approximately 1:100,000 live births. Clinical manifestations and molecular genetic testing are important to confirm the diagnosis. Mutations of the fibroblast growth factor receptor 1 (FGFR1) gene or FGFR2 gene can cause Pfeiffer syndrome. Here, we describe a case of Pfeiffer syndrome with a novel c833_834GC>TG mutation (encoding Cys278Leu) in the FGFR2 gene associated with a coccygeal anomaly, which is rare in Pfeiffer syndrome.
- Introduction
- Introduction
Pfeiffer syndrome is a rare autosomal dominant disorder, characterized by premature fusion of cranial sutures that prevents the skull from growing normally and affects the shape of the head and face, resulting in brachycephaly, hypoplastic maxilla, shallow orbits, proptosis, and exophthalmos, and accompanied with broad and deviated thumbs and big toes1). Molecular genetic testing is important to confirm the diagnosis. Mutations in the fibroblast growth factor receptor (FGFR1) gene2) or FGFR2 gene3) cause Pfeiffer syndrome. Different mutations in the FGFR2 gene have been reported4). Spinal anomalies are rarely associated with Pfeiffer syndrome. Here we describe a case of Pfeiffer syndrome with a novel c833_834GC>TG mutation (encoding Cys278Leu) in the FGFR2 gene, with an everted coccyx.
- Case report
- Case report
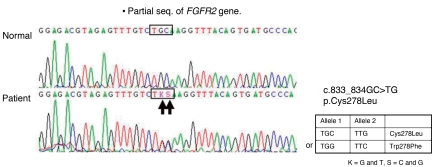
A male infant was born at the 37+3 weeks of gestation via vaginal delivery. His mother was 32 years old, and his father was 31 years old. His mother's obstetric history was gravida 1, para 0, and was unremarkable. His parents were both Korean, phenotypically normal, and did not have any history of consanguinity.The patient's Apgar scores were 9 and 10 at 1 and 5 minute. He weighed 3,360 g (75-90 percentile), was 50 cm long (75 percentile), and had a head circumference of 34 cm (75-90 percentile). He had multiple anomalies such as craniofacial, limbs, and sacral anomalies. Thus he was transferred to Busan Paik Hospital, Inje University College of Medicine. He had brachycephaly, maxillary hypoplasia, exophthalmos, proptosis, low-set ears, preauricular skin tags, atresia of the external auditory canal, high arched palate, radially deviated broad thumbs and medially deviated big toes, ankylosed elbows, and a mass on the coccygeal area (Fig. 1).Plain radiographs of the skull showed prominent convolutions, probably due to craniosynostosis. Three dimensional skull CT revealed closure of the bilateral coronal sutures and left lambdoidal suture, midface hypoplasia, and shallow orbits (Fig. 2A, B). Plain radiographs of his elbows did not show any bony fusion or abnormalities. A lateral spine view noted the disappearance of normal curvature of spine; spinal magnetic resonance imaging showed an outwardly everted coccyx (Fig. 2C), and plain radiographs of his hands and feet showed widening of both 1st phalangeal bones, metacarpal bones, and metatarsal bones. With informed consent, genomic DNA was isolated from peripheral blood. PCR was performed for exon 7 and exon 8 to analyze major mutation of FGFR2 gene, and revealed a novel amino acid substitution of leucine coded by TKS, for cysteine coded by TGC, at codon 833_834 (Cys278Leu) of the FGFR2 gene (Fig. 3).
- Discussion
- Discussion
Pfeiffer syndrome was first described by Pfeiffer in 1964, and 60 cases have been reported. It affects about 1 in 100,000 individuals1), but is more rare in the Asian population, with only a few cases reported in Korea5-8). Pfeiffer syndrome involves the cranial bones and thumbs and great toes, which are broad and bend away from the other digits, occasionally accompanied by an ankylosed elbow or radiohumeral synostosis9).Pfeiffer syndrome is divided into three subtypes9). Type 1 consists of brachycephaly, a hypoplastic mid-face, and finger and toe abnormalities with normal to near-normal intelligence (classic type). Type 2 is characterized by a cloverleaf-shaped head, severe proptosis, and central nervous system involvement caused by more extensive fusion of the skull bones, with potential elbow ankylosis or synostosis. Type 3 is similar to type 2 but without a cloverleaf-shaped head. Both types 2 and 3 are more severe and have poor neurodevelopmental outcomes. The patient in this case report was Pfeiffer syndrome type 1, the classic type.Cranial sutures normally close in a synchronized manner after birth, allowing the skull to achieve normal size and shape. When this normal development is disrupted, premature cranial fusion or delayed cranial closure occurs. Mutations in FGFR1, 2, or 3 can affect craniofacial and skeletal development10). More than 60 mutations in FGFR, a majority of which occur in FGFR2, are associated with craniosynostosis syndrome such as Antley-Bixler syndrome, Apert syndrome, Bearse-Stevenson syndrome, Crouzon syndrome, Muenke syndrome, and Pfeiffer syndrom11). Pfeiffer syndrome is caused by mutations in the FGFR gene with locus heterogeneity. Mutations in the FGFR1 at chromosome 8p11.2-p12 were only detected in Pfeiffer syndrome type 22, 12). Mutations in the FGFR2 at chromosome 10q25-q26 were reported in all three subtypes13), including 1036T->C3) and 1037G->A which were also detected in Crouzon syndrome14). We report a novel c833_834GC>TG mutation (encoding Cys278Leu) in Pfeiffer syndrome. This patient had coccygeal eversion leading to a tail. Spinal anomalies such as the cervical spine fusion are rare in Pfeiffer syndrome15), but coccygeal anomaly occurred in one other case in Korea7).Serum levels of alkaline phosphatase (ALP) increased (1,198 U/L) after birth. 90% of ALP in young infants is produced from bone, and reflects osteoblastic activity, bone turnover, and rapid bone growth, and positively correlates with bone mineral accretion. ALP is normal at birth and rises gradually in the first 2-3 weeks of life16). Gain-of-function mutations in FGFR2 are associated with craniosynostosis. Osteoblast progenitors in bone marrow were significantly increased in Fgfr2cC342Y/+ heterozygote mice, which had a phenotypically shortened face, protruding eyes, and premature fusion of cranial sutures17). Patients with Apert syndrome with the S252W mutation in the FGFR2 gene enhanced osteoblastic marker gene expression and ALP activity18). Increased ALP from birth might reflect prenatally increased osteoblastic marker gene expression resulting from mutations in FGFR2 gene, but further study is needed.The prognosis of Pfeiffer syndrome depends on accompanying anomalies, and multiple surgeries are needed to release the prematurely closed sutures19, 20). In conclusion, this report describes a case of Pfeiffer syndrome with a novel c833_834GC>TG mutation (encoding Cys278Leu) in the FGFR2 gene, with an everted coccyx resembling a tail and increased ALP activity.
- References
- 2. Muenke M, Schell U, Hehr A, Robin NH, Losken HW, Schinzel A, et al. A common mutation in the fibroblast growth factor receptor 1 gene in Pfeiffer syndrome. Nat Genet 1994;8:269–274.
[Article] [PubMed]3. Plomp AS, Hamel BC, Cobben JM, Verloes A, Offermans JP, Lajeunie E, et al. Pfeiffer syndrome type 2: further delineation and review of the literature. Am J Med Genet 1998;75:245–251.
[Article] [PubMed]4. Passos-Bueno MR, Wilcox WR, Jabs EW, Sertie AL, Alonso LG, Kitoh H. Clinical spectrum of fibroblast growth factor receptor mutations. Hum Mutat 1999;14:115–125.
[Article] [PubMed]5. Park MS, Yoo JE, Chung J, Yoon SH. A case of Pfeiffer syndrome. J Korean Med Sci 2006;21:374–378.
[Article] [PubMed] [PMC]6. Chung J, Park DH, Yoon SH. Monoblock craniofacial internal distraction in a child with Pfeiffer syndrome: a case report. J Korean Med Sci 2008;23:342–346.
[Article] [PubMed] [PMC]7. Shin CH, Yi BH, Hong HS, Lee HK, Park SJ. A case report of Pfeiffer syndrome with spinal anomaly. J Korean Soc Radiol 2009;60:57–60.
[Article]8. Yun KW, Rhee KW, Lim IS, Ryu BH. A case of Pfeiffer syndrome with hydrocephalus and multiple congenital anomalies. J Korean Soc Neonatol 2005;12:87–92.9. Cohen MM Jr. Pfeiffer syndrome update, clinical subtypes, and guidelines for differential diagnosis. Am J Med Genet 1993;45:300–307.
[Article] [PubMed]10. Sarkar S, Petiot A, Copp A, Ferretti P, Thorogood P. FGF2 promotes skeletogenic differentiation of cranial neural crest cells. Development 2001;128:2143–2152.
[Article] [PubMed]11. Zhang Y, Gorry MC, Post JC, Ehrlich GD. Genomic organization of the human fibroblast growth factor receptor 2 (FGFR2) gene and comparative analysis of the human FGFR gene family. Gene 1999;230:69–79.
[Article] [PubMed]12. Robin NH, Scott JA, Arnold JE, Goldstein JA, Shilling BB, Marion RW, et al. Favorable prognosis for children with Pfeiffer syndrome types 2 and 3: implications for classification. Am J Med Genet 1998;75:240–244.
[Article] [PubMed]13. Schell U, Hehr A, Feldman GJ, Robin NH, Zackai EH, de Die-Smulders C, et al. Mutations in FGFR1 and FGFR2 cause familial and sporadic Pfeiffer syndrome. Hum Mol Genet 1995;4:323–328.
[Article] [PubMed]14. Reardon W, Winter RM, Rutland P, Pulleyn LJ, Jones BM, Malcolm S. Mutations in the fibroblast growth factor receptor 2 gene cause Crouzon syndrome. Nat Genet 1994;8:98–103.
[Article] [PubMed]15. Moore MH, Lodge ML, Clark BE. Spinal anomalies in Pfeiffer syndrome. Cleft Palate Craniofac J 1995;32:251–254.
[Article] [PubMed]16. Harrison CM, Johnson K, McKechnie E. Osteopenia of prematurity: a national survey and review of practice. Acta Paediatr 2008;97:407–413.
[Article] [PubMed]17. Eswarakumar VP, Horowitz MC, Locklin R, Morriss-Kay GM, Lonai P. A gain-of-function mutation of Fgfr2c demonstrates the roles of this receptor variant in osteogenesis. Proc Natl Acad Sci U S A 2004;101:12555–12560.
[Article] [PubMed] [PMC]18. Tanimoto Y, Yokozeki M, Hiura K, Matsumoto K, Nakanishi H, Matsumoto T, et al. A soluble form of fibroblast growth factor receptor 2 (FGFR2) with S252W mutation acts as an efficient inhibitor for the enhanced osteoblastic differentiation caused by FGFR2 activation in Apert syndrome. J Biol Chem 2004;279:45926–45934.
[Article] [PubMed]
Fig. 1
Photographs of the patient. (A) Brachycephaly, hypoplastic mid-face, proptosis, and exophthalmos are shown in the photograph of the head. (B, C) The thumbs and the great toes are wide and bend away from the other digits.
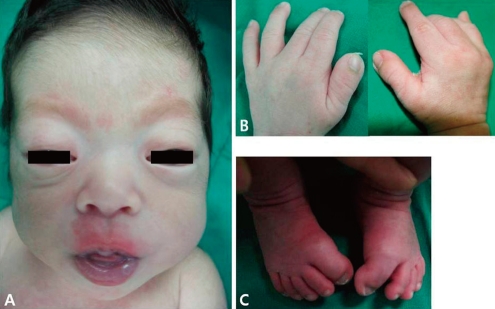
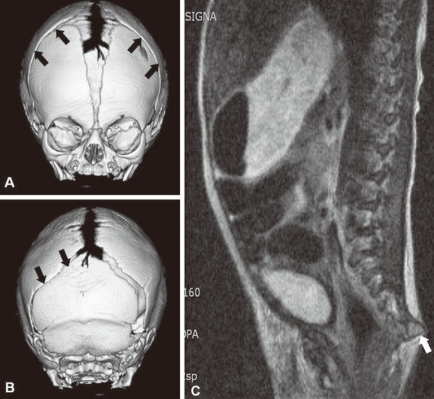
Fig. 2
Three-dimensional skull computed tomography (CT) and spine magnetic resonance imaging. A, B) Three dimensional CT shows closure of bilateral coronal sutures (arrows) and left lambdoidal suture (arrows) with sclerotic changes, and hypoplastic facial bone and shallow orbit. C) Spine magnetic resonance imaging shows an outwardly everted coccyx (arrow) and the disappearance of normal vertebral curvature.