About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 53(10); 2010
The neuroprotective effect of recombinant human erythropoietin via an antiapoptotic mechanism on hypoxic-ischemic brain injury in neonatal rats
- Corresponding author: Woo Taek Kim, M.D. Department of Pediatrics, School of Medicine, Catholic University of Daegu, #3056-6, Daemyung-4-dong, Nam-gu, Daegu 705-718, Korea. Tel: +82.53-650-4250, Fax: +82.53-622-4240, wootykim@cu.ac.kr
- Received February 16, 2010 Revised May 17, 2010 Accepted July 20, 2010
- Abstract
-
- Purpose
- Purpose
- The neuroprotective effects of erythropoietin (EPO) have been recently shown in many animal models of brain injury, including hypoxic-ischemic (HI) encephalopathy, trauma, and excitotoxicity; however, limited data are available for such effects during the neonatal periods. Therefore, we investigated whether recombinant human EPO (rHuEPO) can protect against perinatal HI brain injury via an antiapoptotic mechanism.
- Methods
- Methods
- The left carotid artery was ligated in 7-day-old Sprague-Dawley (SD) rat pups (in vivo model). The animals were divided into 6 groups: normoxia control (NC), normoxia sham-operated (NS), hypoxia only (H), hypoxia+vehicle (HV), hypoxia+rHuEPO before a hypoxic insult (HE-B), and hypoxia+rHuEPO after a hypoxic insult (HE-A). Embryonic cortical neuronal cell culture of SD rats at 18 days gestation (in vitro model) was performed. The cultured cells were divided into 5 groups: normoxia (N), hypoxia (H), and 1, 10, and 100 IU/mL rHuEPO-treated groups.
- Results
- Results
- In the in vivo model, Bcl-2 expressions in the H and HV groups were lower than those in the NC and NS groups, whereas those in the HE-A and HE-B groups were greater than those of the H and HV groups. The expressions of Bax and caspase-3 and the ratio of Bax/Bcl-2 were in contrast to those of Bcl-2. In the in vitro model, the patterns of Bcl-2, Bax, and caspase-3 expression and Bax/Bcl-2 ratio were similar to the results obtained in the in vivo model.
- Conclusion
- Conclusion
- rHuEPO exerts neuroprotective effect against perinatal HI brain injury via an antiapoptotic mechanism.
- Introduction
- Introduction
Erythropoietin (EPO), a hematopoietic cytokine known best for its erythropoietic effects, has neuroprotective effects in many animal models of brain injury, including hypoxic-ischemic (HI) encephlopathy, trauma and excitotoxicity1). The biological effects of EPO in the central nervous system (CNS) are mediated by binding to its specific receptor (EPO-R). The expression of EPO and EPO-R in the nervous system is modulated by hypoxia and metabolic insult2). EPO-R expression is abundant in the embryonic, fetal and adult brain of rats, mice, monkeys and humans3, 4). EPO has been identified as a neurotrophic and neuroprotective agent in a wide variety of experimental paradigms, from neuronal cell culture to in vivo models of brain injury. Although the exact mechanisms of the neuroprotective action of EPO are not completely known, promotion of cell survival signaling cascades, modulation of intracellular calcium metabolism, attenuation of nitric oxide (NO) production, inhibition of glutamate release, anti-apoptosis, antioxidative and anti-inflammatory actions have been suggested as possible mechanisms5).EPO is known to promote cell survival via Bcl-2 family of genes in non-neuronal tissues and regulates the expression of apoptosis-related genes from Bcl-2 family in different experimental paradigms6). EPO is able to shift the Bcl/Bax ratio towards a net antiapoptotic effect in cultured microglia7). It also upregulates Bcl-xl mRNA and protein expressions in cultured neurons8). EPO reduced retinal ganglion cell death induced by glutamate and NO, and reversed Bcl-2 downregulation9). EPO also increases Bcl-2 gene expression in rat cardiac myocytes after traumatic brain injury10).Various studies have demonstrated a protective effect of EPO in models of brain injury in vitro and in vivo7, 10, 11). Here we investigated whether recombinant human EPO (rHuEPO) can protect the developing rat brain from HI injury via an anti-apoptosis analyzing possible molecular mechanisms accounting for the neuroprotective effect of rHuEPO.
- Materials and methods
- Materials and methods
- 1. Materials
- 1. Materials
rHuEPO was purchased from CJ Cheiljedang Corporation (Seoul, Korea). Primary antibodies included the following: Bcl-2 (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), Bax (1:1,000; Cell Signaling technolog, Danvers, MA, USA) and caspase-3 (1:1,000; Cell Signaling technology). Secondary antibodies used were goat anti-mouse or rabbit IgG-HRP (1:2000, Santa Cruz Biotechnology).- 2. Animal protocol and drug administration
- 2. Animal protocol and drug administration
The neonatal rat HI procedure was performed as described by Rice et al.12). We chose 7-day-old Sprague-Dawley (SD) rat pups weighing between 12 and 16 g. The rat pups were anesthetized with ether. The left common carotid artery (CCA) was exposed, isolated and permanently doubly ligated with 5-0 surgical silk. The incision was sutured and the animal was allowed to recover in a warm environment. The whole surgical procedure was completed in less than 5 minutes. After a recovery period of 1 hour, the pups were placed in plastic chambers, which were submerged in a 37℃ water bath and exposed to hypoxia (mixture of 8% O2 and 92% N2) for 2 hours. After this hypoxic exposure, the pups were returned to their dams for the indicated time.rHuEPO was prepared in phosphate-buffered saline (PBS) and injected intraperitoneally at a dose of 1,000 IU/kg 30 minutes before or after a hypoxic insult.The animals were divided into six groups. In group 1 (Normoxia control, NC, n=7), they were not exposed to hypoxia. In group 2 (Normoxia Sham-operated, NS, n=8), they were subjected only to open-close surgery without CCA ligation. In group 3 (Hypoxia only, H, n=8), they were subjected to hypoxia without rHuEPO injection. In group 4 (Hypoxia+vehicle, HV, n=7), they were injected with PBS (the same volume of rHuEPO). In group 5 (Hypoxia+rHuEPO "before a hypoxic insult", HE-B, n=8), they were injected with 1,000 IU/kg of rHuEPO 30 minutes before a hypoxic insult. In group 6 (Hypoxia+rHuEPO "after a hypoxic insult", HE-A, n=8), they were injected with the same dose of rHuEPO 30 minutes after a hypoxic insult. Pups from each litter were randomly assigned and marked to the NC, NS, H, HV, HE-B or HE-A groups.- 3. Hematoxylin and eosin (H & E) staining
- 3. Hematoxylin and eosin (H & E) staining
Histologic studies were performed 7 days after a HI insult. After intracardiac perfusion with saline, the brains were removed, fixed in 4% paraformaldehyde, embedded in paraffin, and prepared for light microscopy. Four µm sections were mounted on glass cover slides. Sections were deparaffinized in xylene for 30 minutes and serially treated with 100% (5 minutes), 96% (10 minutes), and 70% (10 minutes) ethanol. Slides were stained with hematoxylin, rinsed for a few seconds with water to remove excess stain, placed in 1% eosin, rinsed briefly in water to remove the acid and washed through the following series: 70%, 95% and 100% ethanol at 5 minutes each. Finally, the slides were transferred to xylene for clearing, mounted, and covered with cover slips.Each slides were taken a photograph and were measured brain area with a densitometer (Multi-gauze software, Fuji Photofilm, Tokyo, Japan).- 4. Cortical neuronal cell culture of rat embryos
- 4. Cortical neuronal cell culture of rat embryos
The cortical neuronal cell culture of rat embryos based on the Brewer13) method was performed. SD rats pregnant for 19 days were anesthetized with ether for 5 minutes at room temperature and the uteri were removed. The fetal pups were washed in 100% ethanol and Hanks' balanced salt solution (HBSS) (GibcoBRL, Grand Island, NY, USA). The fetal pup brains were dissected at 37℃ HBSS containing 1 mM sodium pyruvate and 10 mM HEPES (pH 7.4). The dissected brain cortical tissues were then placed in 2 mL trypsin and incubated at 37℃ water bath for 1 minute. Trypsin was removed by washing the tissues five times with 10 mL HBSS. The cells were moved in 1 mL HBSS, passed 6-7 times via pasteur pipette with tiny hole and dispersed. The cell suspension was centrifuged at 1,000 rpm at 25℃ for 5 minutes and pellets were washed with HBSS (without phenol red).After cell counting, they were inoculated in plating Neurobasal media (GibcoBRL) (100 mL neurobasal, 2 mL B27 supplement, 0.25 mL glutamax I, 0.1 mL 25 mM glutamate, 0.1 mL 25 mM 2-mercaptoethanol) with about 2×106 cells/mm2 in each dish. The cell cultures were incubated in a CO2 chamber. A fifth of the culture solutions were changed every three days and were replaced with feeding Neurobasal media (GibcoBRL).The cultured cells were divided into five groups: normoxia group (N), hypoxia group (H) and hypoxia+1, 10 and 100 IU/mL rHuEPO groups (H+E1, H+E10 and H+E100). The N group was prepared in 5% CO2 incubators while the H group and H+E groups in 1% O2 incubators (94% N2, 5% CO2) for 6 hours. The experiments were repeated four times (n=4).- 5. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay
- 5. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay
The MTT assay was used for estimation of cell viability and growth as originally described by Mosmann17). MTT was dissolved at a concentration of 5 mg/mL. Ten µL of the 5 mg/mL MTT stock solution was added to each well. After 4 hour of incubation at 37℃, media was removed and added 100 µL of the lysing buffer (Dimethyl sulfoxide (DMSO) : 95% ethanol = 1 : 1). Absorbance of the samples was read at 492 nm using a microtiter plate enzyme-linked immunosorbent assay (ELISA) reader. The amount of formazan produced is proportional to the number of live and metabolically active cells.- 6. Brain extraction and protein isolation
- 6. Brain extraction and protein isolation
Pups were killed 7 days after a hypoxic insult. Left cerebral hemispheres from rat brains were immediately removed, frozen in liquid nitrogen and stored at -70℃ until use. Frozen tissues were homogenized in protein lysis buffer containing complete protease inhibitor cocktail tablets (Roche Applied Science, Mannheim, Germany), 1 M Tris-HCl (pH 8.0), 5 M NaCl, 10% Nonidet P-40 and 1 M 1,4-dithio-DL-threitol (DTT). After incubation for 20 minutes on ice, the samples were centrifuged at 12,000 rpm at 4℃ for 30 minutes and the supernatant was transferred to a new tube. Total protein was measured with a Bio-Rad Bradford kit (Bio-rad Laboratories, Hercules, CA, USA).- 7. Western blotting
- 7. Western blotting
Samples of equal amounts of proteins (30 µg) were subjected to a 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) after denaturing in SDS gel-loading buffer (100 mM Tris-HCl (pH 6.8), 200 mM DTT, 20% glycerol, 4% SDS and 0.2% bromophenol blue) in boiling water for 10 minutes. After electrophoresis proteins were electrotransferred onto polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA, USA) at a constant voltage of 10 V for 30 minutes. After transfer, the membrane was washed twice with 1 × tris-buffered saline (TBS) plus 0.1% Tween-20 (TBST pH 7.4) and then pre-incubated with a blocking buffer (5% nonfat dry milk in TBST) at room temperature for 1 hour. The membrane was then incubated with the primary antibodies 1:1,000 dilutions in TBST at 4℃ overnight. After washing, the blots were incubated with the secondary antibodies (1:2,000 dilution) at room temperature for 1 hour. Finally, the membrane was washed and developed with an enhanced chemiluminoescence (ECL) Plus Western Blotting Detection System (Amersham Biosciences, Piscataway, NJ, USA) or SUPEX (Neuronex, Pohang, Korea).- 8. Semiquantitation of the western blots
- 8. Semiquantitation of the western blots
The intensity of the corresponding western blot band was measured by using a densitometer (Multi Gauge Software) and was calculated as the ratio of the signal intensity in the ischemic (left) hemisphere compared to the contralateral (right) hemisphere.- 9. RNA extraction and real-time PCR
- 9. RNA extraction and real-time PCR
Total RNA extraction was carried out with TRIzol reagent (Invitrogen Corporation, Carlsbad, CA, USA). Briefly, a piece of tissue was homogenized in 1 mL of TRIzol reagent. Total RNA was separated from DNA and proteins by adding chloroform and was precipitated using isopropanol. The precipitate was washed twice in 100% ethanol, air-dried and re-diluted in diethylpyrocarbonate (DEPC)-treated distilled water. The amount and purity of extracted RNA was quantitated by GeneQuant 1,300 spectrophotometer (GE Healthcare, UK). The RNA was then stored at -70℃ before further processing. For real-time PCR (for reverse transcription), total RNA (1 µg) was reversely transcribed for 1 hour at 37℃ in a reaction mixture containing 20 U RNase inhibitor (Promega, Madison, WI), 1 mM dNTP (Promega, Madison, WI, USA), 0.5 ng Oligo (dT) 15 primer (Promega, Madison, WI, USA), 1 × RT buffer and 200 U M-MLV reverse transcriptase (Promega, Madison, WI, USA). The reaction mixture was then incubated at 95℃ for 5 minutes to stop the reaction. The cDNA was then stored at -20℃ before further processing.Real-time PCR was performed in 48 well PCR plates (Mini OpticonTM Real-Time PCR System) using the FINNZYMES DyNAmo SYBR green qPCR kit (Finnzymes, Beverly, MA, USA). Amplification conditions are shown in Table 1. The thermal profile was 95℃ for 15 minutes followed by 40 cycles of denaturation, annealing and extension as indicated in Table 1. Real-time PCR data were analysed with LightCycler software (Bio-rad Laboratories, Hercules, CA, USA). Each sample was conducted in triplicate.- 10. Statistics analysis
- 10. Statistics analysis
Data were analyzed using the SPSS version 12 statistical analysis package (SPSS Inc., Chicago, IL, USA). Examined data were assessed using the t-test, GLM (general lineal model), and ANOVA. In each test, the data were expressed as the mean±SD, and P<0.05 was accepted as statistically significant.
- Results
- Results
- 1. H & E staining after a hypoxic insult of neonatal rat brain
- 1. H & E staining after a hypoxic insult of neonatal rat brain
H & E stain of 7-day-old rat brain after a HI insult revealed that the brain areas in the H (C, 58.5%) and HV (D, 68.6%) groups were decreased compared to the NC (A, 102.8%) and NS (B, 94.8%) groups, whereas those in the HE-B (E, 94.3%) and HE-A (F, 81.4%) groups were increased compared to the H (C, 58.5%) and HV (D, 68.6%) groups (Fig. 1). Percentages were the ratios of left hemisphere area compared to right hemisphere area.- 2. The expressions of Bcl-2, Bax and caspase-3 proteins by western blot analysis in the neonatal HI brain injury
- 2. The expressions of Bcl-2, Bax and caspase-3 proteins by western blot analysis in the neonatal HI brain injury
The expressions of Bcl-2 in the H and HV groups were decreased compared to the NC and NS groups, whereas those in the HE-A and HE-B groups were increased compared to the H and HV groups (P<0.05) (Fig. 2A). The expressions of Bax and caspase-3 and the ratio of Bax/Bcl-2 in the H and HV groups were increased compared to the NC and NS groups, whereas those in the HE-A and HE-B groups were decreased compared to the H and HV groups (P<0.05) (Fig. 2B, 2C and 2D).- 3. The expressions of Bcl-2, Bax and caspase-3 mRNAs by real-time PCR on the HI brain injury in neonatal rats
- 3. The expressions of Bcl-2, Bax and caspase-3 mRNAs by real-time PCR on the HI brain injury in neonatal rats
The expressions of Bcl-2 in the H and HV groups were decreased compared to the NC and NS groups, whereas those in the HE-A and HE-B groups were increased compared to the H and HV groups (P<0.05) (Fig. 3A). The expressions of Bax and caspase-3 and the ratio of Bax/Bcl-2 in the H and HV groups were increased compared to the NC and NS groups, whereas those in the HE-A and HE-B groups were decreased compared to the H and HV groups (P<0.05) (Fig. 3B, 3C and 3D).- 4. The images of cortical neuronal cell culture of rat embryos
- 4. The images of cortical neuronal cell culture of rat embryos
The cortical neuronal cells were observed using light microscopy under high magnification (×200). The cells in the N group (Fig. 4A) appeared normal, whereas those in the H group (Fig. 4B) showed cellular damages. The cellular patterns of the H+E1 (Fig. 4C) and H+E10 group (Fig. 4D) were similar to those in the N group, whereas the cells in the H+E100 group (Fig. 4E) appeared similar to those in the H group.- 5. Cell viabilities of cultured cortical neuronal cells determined by the MTT assay
- 5. Cell viabilities of cultured cortical neuronal cells determined by the MTT assay
The relative cell viabilities in the H group were decreased compared to the N group. The relative cell viabilities in the H+E1 and H+E10 group were similar to those in the N group, whereas those in the H+E100 group appeared similar to those in the H group (Fig. 5).- 6. The expressions of Bcl-2, Bax and caspase-3 by proteins western blot analysis in the cortical neuronal cell culture
- 6. The expressions of Bcl-2, Bax and caspase-3 by proteins western blot analysis in the cortical neuronal cell culture
The expressions of Bcl-2 in the H group was decreased compared to the N group. whereas those in the H+E10 group were increased compared to the H group (P<0.05) (Fig. 6A). The expressions of Bax and the ratio of Bax/Bcl-2 in the H group were increased compared to the N group, whereas those in the H+E10 group were decreased gradually depending to the concentration of rHuEPO (P<0.05) (Fig. 6B and 6D). The expressions of caspase-3 the in the H group were increased compared to the N group, whereas those in the H+E1 and H+E10 groups were decreased compared to the H group (P<0.05) (Fig. 6C).- 7. The expressions of Bcl-2, Bax and caspase-3 mRNA by real-time PCR in cortical neuronal cell culture
- 7. The expressions of Bcl-2, Bax and caspase-3 mRNA by real-time PCR in cortical neuronal cell culture
The expressions of Bcl-2 in the H group were decreased compared to the N group. whereas those in the H+E1 group was increased compared to the H group (P<0.05) (Fig. 7A). The expressions of Bax and the ratio of Bax/Bcl-2 in the H group were increased compared to the N group, whereas those in the H+E1, H+E10 and H+E100 groups were decreased compared to the H group (P<0.05) (Fig. 7B and 7D). The expressions of caspase-3 in the H group were increased compared to the N group, whereas those in the H+E1 and H+E10 groups were decreased compared to the H group (P<0.05) (Fig. 7C).
- Discussion
- Discussion
HI brain injury in the human perinatal period can result from several pathological processes such as premature placental detachment or other complications of pregnancy and delivery. It is an important cause of neonatal mortality and subsequent sequelae such as cerebral palsy, mental retardation, learning disability and epilepsy15). Hypoxia, ischemia, and more recently, intrauterine and neonatal infections, have been implicated in the pathogenesis of injury to the developing brain15). Neonates who were exposed to HI brain injury sustain both acute and chronic morbidities that can affect the sensory, motor, cognitive and behavioral systems. The full extent of this injury may not be apparent until the child reaches school age or later5). Premature neonates are particularly vulnerable to HI exposure and may develop a variety of deficits that will reveal themselves when tested at school entry16).A variety of therapeutic strategies are employed to protect against the pathologic mechanisms of HI injury. These include induction of hypothermia, antagonism of excitotoxic glutamate receptors, anti-inflammatory agents, neurotropic factors and inhibitors of the caspase pathway and of stress kinases5). Pharmacological agents which mediate one or more of these processes may provide neuroprotection for patients against this condition. Furthermore, study of neuroprotective agents may provide important information about treatment for the perinatal HI brain injury. The in vivo neonatal rat HI model and the in vitro cortical cell culture model of rat embryos have been well characterized and used extensively to search for neuroprotective agents17).EPO, a 34-kDa glycoprotein hormone, was first characterized as a hematopoietic growth factor. It is now widely used for the treatment of anemia associated with renal failure, cancer, prematurity, chronic inflammatory disease and human immunodeficiency virus infection. EPO is initially produced in the liver but shortly after birth, its production is shifted to the kidney18). EPO has been originally characterized as the principal regulator of erythropoiesis by inhibiting apoptosis and by stimulating the proliferation and differentiation of erythroid precursor cells19). EPO exerts remarkable neuroprotection in both cell cultures and in animal models of nervous system disorders and has been implicated in the development of the CNS. Neuroprotective effect of EPO has also been demonstrated on various animal models of cerebral ischemia20).Recent studies have shown that EPO protects neonatal brains as well from HI injury by reducing apoptotic cell death in vivo21) and protects cultured neurons against oxygen glucose deprivation injury by a paracrine protective pathway through astrocytes expressing EPO22). EPO is also found to protect the developing rat brain against NMDA receptor antagonist neurotoxicity and protects the mouse retina against light-induced degeneration by inhibiting photoreceptor cell apoptosis23). Our in vivo experimental study revealed that the brain areas in hypoxia were decreased compared to normoxia, whereas those in EPO-treated groups were increased compared to hypoxia.In in vitro experiments, EPO has been shown to protect cultured hippocampal and cortical neurons against glutamate toxicity24), hypoxia and glucose deprivation25) and serum deprivation-induced and kainic acid-induced apoptosis24). In addition, systemic administrations of rHuEPO as a pretreatment or posttreatment have been shown to effectively reduce brain injury after focal ischemia26), subarachnoid hemorrhage27) and HI28). Our in vitro experimental study showed the cells in normoxia appeared normal, whereas those in hypoxia showed cellular damages. And the cellular patterns in the low dose EPO-treated groups were similar to those in normoxia, whereas the cells in the high dose EPO-treated groups appeared similar to those in hypoxia.Both EPO and its specific receptor (erythropoietin receptor, EPO-R) are expressed in different tissues, including the nervous system19). EPO acts via activation of the specific EPO-R. EPO and EPO-R are present in the developing human brain as early as 5 weeks post-conception, and each protein shows a specific distribution that changes with development29). EPO-R have been identified in the nervous system of rodents, primates and humans19, 29) and have been localized on several primary cell lines of neurons, as well as in glial cells. It has been shown that EPO stimulates neurogenesis and prevents apoptosis in the embryonic brain30). EPO-R is widespread throughout fetal brain29). EPO-R binding initiates a signaling cascade that involves multiple antiapoptotic pathways that can account for the rHuEPO-induced reduction26). EPO-R activation protects hippocampal neurons from ischemic neuronal damage through the inhibition of glutamate release via exocytosis in hippocampal slice cultures. In the brain, EPO is released by astrocytes22), triggers dopamine release31), promotes neurogenesis32) and vasculogenesis33), and stimulates proliferation of glia34). EPO is therefore recognized to have both trophic and antiapoptotic mechanisms of action.EPO might have a direct protective role against a variety of neurotoxic insults such as hypoxic conditions24), glutamate toxicity19), free-radical injury35) and exposure to neurotoxicants36). EPO stimulates dopamine release and tyrosine hydroxylase activity in PC12 cells, through the activation of calcium channels, induces membrane depolarization, stimulates mitogen activated protein kinase activity and increases NO synthesis37). EPO reduces calcium-induced glutamate release from cultured cerebellar granule neurons38).Apoptosis contributes significantly to cerebral damage during the perinatal period. In neonatal HI model, there is evidence that cerebral ischemia leads to delayed cell death with DNA damage39). EPO has anti-apoptotic action on some non-neural cells40). There is some evidence suggesting that EPO may also protect neuronal cells that morphologically die via apoptosis both in vivo and in vitro. And EPO may also exert its neuroprotective effect via the differential regulating of gene expression ruling the balance between the expression of pro- and anti-apoptotic molecules. EPO prevents ischemia-induced delayed neuronal death in the hippocampus in animal model of global cerebral ischemia11). EPO decreases apoptotic cell death of neurons under conditions of hypoxia in vitro, suggesting that it might have similar effect in vivo29). Antiapoptotic action of EPO has also been confirmed in focal cerebral ischemia and transient spinal ischemia models24). EPO decreases apoptotic cell death of neurons under conditions of hypoxia in vitro , suggesting that it might have a similar effect in vivo29).EPO inhibits caspase 8-, caspase 1- and caspase 3-like activities linked to cytochrome c release35). EPO consistently increases the expression of the anti-apoptotic gene Bcl-XL and decreases the expression of pro-apoptotic gene bak in PC12 cells. EPO is able to shift the Bcl/Bax ratio towards a net anti-apoptotic effect in cultured microglia7). It also up-regulates Bcl-XL mRNA and protein expressions in cultured neurons8). EPO rescued hippocampal CA1 neurons from lethal ischemic damage and up-regulated the expressions of Bcl-XL mRNA and protein in the hippocampal CA1 field of ischemic gerbils. Thus, EPO prevents delayed neuronal death of hippocampal neurons, possibly through up-regulation of Bcl-XL, which is known to facilitate neuronal survival8).Western blot analysis and real-time PCR both our in vitro and in vivo models of perinatal HI brain injury revealed that the expressions of Bcl-2 in hypoxia were decreased compared to in nomoxia whereas those in rHuEPO-treated groups were increased compared to in hypoxia. The expressions of Bax after hypoxia as well as the ratio of Bax/Bcl-2 in hypoxia were increased compared to nomoxia whereas those in rHuEPO-treated groups decreased compared to hypoxia. In addition, the expressions of caspase-3 in hypoxia were increased compared to nomoxia whereas those in rHuEPO-treated groups were decreased compared to nomoxia.The above data suggest that rHuEPO's nuroprotective effect against neonatal HI brain injury may have been via an anti-apoptosis mechanism which the ratio of Bax/Bcl-2 and the expressions of caspase-3 in rHuEPO-treated groups were increased compared to hpoxia.In conclusion, our experiments demonstrate that rHuEPO is able to prevent the degeneration of neonatal cerebral neuronal cells caused by a hypoxic insult. In addition, rHuEPO's neuroprotective effects on HI brain injury in neonatal rats may be via anti-apoptosis. The present study may be useful for the further development of clinical therapies for perinatal HI encephalopathy-induced by cerebral hypoxia.
- Acknowledgments
This work was supported by the grant of Research Institute of Medical Science, Catholic University of Daegu(2009)
- References
- 1. Liu R, Suzuki A, Guo Z, Mizuno Y, Urabe T. Intrinsic and extrinsic erythropoietin enhances neuroprotection against ischemia and reperfusion injury in vitro. J Neurochem 2006;96:1101–1110.
[Article] [PubMed]2. Dame C, Juul S, Christensen RD. The biology of erythropoietin in the central nervous system and its neurotrophic and neuroprotective potential. Biol Neonate 2001;79:228–235.
[Article] [PubMed]3. Digicaylioglu M, Bichet S, Marti HH, Wenger RH, Rivas LA, Bauer C, et al. Localization of specific erythropoietin binding sites in defined areas of the mouse brain. Proc Natl Acad Sci USA 1995;92:3717–3720.
[Article] [PubMed] [PMC]4. Marti HH, Wenger RH, Rivas LA, Straumann U, Digicaylioglu M, Hehn V, et al. Erythropoietin gene expression in human monkey and murine brain. Eur J Neurosci 1996;8:666–676.
[Article] [PubMed]5. Volpe JJ. Perinatal brain injury: from pathogenesis to neuroprotection. Ment Retard Dev Disabil Res Rev 2001;7:56–64.
[Article] [PubMed]6. Chong ZZ, Kang JQ, Maiese K. Hematopoietic factor erythropoietin fosters neuroprotection through novel signal transduction cascades. J Cereb Blood Flow Metab 2002;22:503–514.
[Article] [PubMed]7. Vairano M, Russo CD, Pozzoli G, Battaglia A, Scambia G, Tringali G, et al. Erythropoietin exerts anti-apoptotic effects on rat microglial cells in vitro. Eur J Neurosci 2002;16:584–592.
[Article] [PubMed]8. Wen TC, Sadamoto Y, Tanaka J, Zhu PX, Nakata K, Ma YJ, et al. Erythropoietin protects neurons against chemical hypoxia and cerebral ischemic injury by upregulating Bcl-xL expression. J Neurosci Res 2002;67:795–803.
[Article] [PubMed]9. Yamasaki M, Mishima HK, Yamashita H, Kashiwagi K, Murata K, Minamoto A, et al. Neuroprotective effects of erythropoietin on glutamate and nitric oxide toxicity in primary cultured retinal ganglion cells. Brain Res 2005;1050:15–26.
[Article] [PubMed]10. Emir M, Ozisik K, Cagli K, Misirlioglu M, Ozisik P, Iscan Z, et al. Effect of erythropoietin on bcl-2 gene expression in rat cardiac myocytes after traumatic brain injury. Transplant Proc 2004;36:2935–2938.
[Article] [PubMed]11. Sakanaka M, Wen TC, Matsuda S, Masuda S, Morishita E, Nagao M, et al. In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc Natl Acad Sci USA 1998;95:4635–4640.
[Article] [PubMed] [PMC]12. Rice JE 3rd, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol 1981;9:131–141.
[Article] [PubMed]13. Brewer GJ. Isolation and culture of adult rat hippocampal neurons. J Neurosci Methods 1997;71:143–155.
[Article] [PubMed]14. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63.
[Article] [PubMed]15. Vannucci RC, Perlman JM. Interventions for perinatal hypoxic-ischemic encephalopathy. Pediatrics 1997;100:1004–1014.
[Article] [PubMed]16. Anderson P, Doyle LW. Victorian Infant Collaborative Study Group. Neurobehavioral outcomes of school-age children born extremely low birth weight or very preterm in the 1990s. JAMA 2003;289:3264–3272.
[Article] [PubMed]17. Feng Y, Fratkin JD, LeBlanc MH. Treatment with tamoxifen reduces hypoxic-ischemic brain injury in neonatal rats. Eur J Pharmacol 2004;484:65–74.
[Article] [PubMed]18. Jelkmann W. Erythropoietin: structure, control of production, and function. Physiol Rev 1992;72:449–489.
[Article] [PubMed]19. Siren AL, Fratelli M, Brines M, Goemans C, Casagrande S, Lewczuk P, et al. Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc Natl Acad Sci USA 2001;98:4044–4049.
[Article] [PubMed] [PMC]20. Bernaudin M, Marti HH, Roussel S, Divoux D, Nouvelot A, MacKenzie ET, et al. A potential role for erythropoietin in focal permanent cerebral ischemia in mice. J Cereb Blood Flow Metab 1999;19:643–651.
[Article] [PubMed]21. Spandou E, Soubasi V, Papoutsopoulou S, Karkavelas G, Simeonidou C, Kaiki-Astara A, et al. Erythropoietin prevents hypoxia/ischemia-induced DNA fragmentation in an experimental model of perinatal asphyxia. Neurosci Lett 2004;366:24–28.
[Article] [PubMed]22. Ruscher K, Freyer D, Karsch M, Isaev N, Megow D, Sawitzki B, et al. Erythropoietin is a paracrine mediator of ischemic tolerance in the brain, evidence from an in vitro model. J Neurosci 2002;22:10291–10301.
[Article] [PubMed] [PMC]23. Dzietko M, Felderhoff-Mueser U, Sifringer M, Krutz B, Bittigau P, Thor F, et al. Erythropoietin protects the developing brain against N-methyl-d-aspartate receptor antagonist neurotoxicity. Neurobiol Dis 2004;15:177–187.
[Article] [PubMed]24. Morishita E, Masuda S, Nagao M, Tasuda Y, Sasaki R. Erythropoietin receptor is expressed in rat hippocampal and cerebral cortical neurons, and erythropoietin prevents in vitro glutamateinduced neuronal death. Neuroscience 1997;76:105–116.
[Article] [PubMed]25. Sinor AD, Greenberg DA. Erythropoietin protects cultured cortical neurons, but not astroglia, from hypoxia and AMPA toxicity. Neurosci Lett 2000;290:213–215.
[Article] [PubMed]26. Brines M, Cerami A. Emerging biological roles for erythropoietin in the nervous system. Nat Rev Neurosci 2005;6:484–494.
[Article] [PubMed]27. Grasso G, Sfacteria A, Cerami A, Brines M. Erythropoietin as a tissueprotective cytokine in brain injury: what do we know and where do we go? Neuroscientist 2004;10:93–98.
[Article] [PubMed]28. Ghezzi P, Brines M. Erythropoietin as an antiapoptotic, tissue-protective cytokine. Cell Death Differ 2004;11:S37–S44.
[Article] [PubMed]29. Juul SE, Yachnis AT, Rojiani AM, Christensen RD. Immunohistochemical localization of erythropoietin and its receptor in the developing human brain. Pediatr Dev Pathol 1999;2:148–158.
[Article] [PubMed]30. Yu X, Shacka JJ, Eels JB, Suarez-Quinan C, Przygodski RM, Beleslin-Cokic B, et al. Erythropoietin receptor signalling is required for normal brain development. Development 2002;129:505–516.
[Article] [PubMed]31. Markianos M, Kosmidis ML, Sfagos C. Reductions in plasma prolactin during acute erythropoietin administration. Neuro Endocrinol Lett 2006;27:355–358.
[PubMed]32. Wang L, Zhang Z, Wang Y, Zhang R, Chopp M. Treatment of stroke with erythropoietin enhances neurogenesis and angiogenesis and improves neurological function in rats. Stroke 2004;35:1732–1737.
[Article] [PubMed]33. Ribatti D, Vacca A, Roccaro AM, Crivellato E, Presta M. Erythropoietin as an angiogenic factor. Eur J Clin Invest 2003;33:891–896.
[Article] [PubMed]34. Sugawa M, Sakurai Y, Ishikawa-Ieda Y, Suzuki H, Asou H. Effects of erythropoietin on glial cell development; oligodendrocyte maturation and astrocyte proliferation. Neurosci Res 2002;44:391–403.
[Article] [PubMed]35. Chong ZZ, Lin SH, Kang JQ, Maiese K. Erythropoietin prevents early and late neuronal demise through modulation of Akt1 and induction of caspase 1, 3, and 8. J Neurosci Res 2003;71:659–669.
[Article] [PubMed]36. Villa P, Bigini P, Mennini T, Agnello D, Laragione T, Cagnotto A, et al. Erythropoietin selectively attenuates cytokine production and inflammation in cerebral ischemia by targeting neuronal apoptosis. J Exp Med 2003;198:971–975.
[Article] [PubMed] [PMC]37. Koshimura K, Murakami Y, Sohmiya M, Tanaka J, Kato Y. Effects of erythropoietin on neuronal activity. J Neurochem 1999;72:2565–2572.
[Article] [PubMed]38. Kawakami M, Sekiguchi M, Sato K, Kozaki S, Takahashi M. Erythropoietin receptor-mediated inhibition of exocytotic glutamate release confers neuroprotection during chemical ischemia. J Biol Chem 2001;276:39469–39475.
[Article] [PubMed]
Fig. 1
Hematoxylin and eosin (H&E) staining after hypoxic insult of neonatal rat brain. A, Normoxia control group; B, Normoxia sham-operated group; C, Hypoxia group; D, Hypoxia+Vehicle group; E, Hypoxia+Recombinant human erythropoietin (rHuEPO)-treated group before hypoxic insult; F, Hypoxia+rHuEPO-treated group after hypoxic insult. Comparison of the percentage of the area of the left hemisphere area with that of the right hemisphere: A, 102.8%; B, 94.8%; C, 58.5%; D, 68.6%; E, 94.3%; and F, 81.4%.
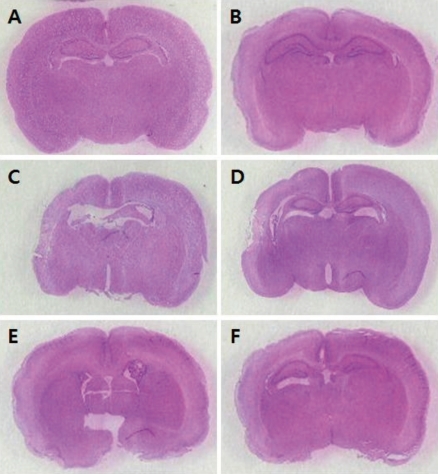
Fig. 2
Western blots of Bcl-2 (A, NC, 100±0; NS, 104.8±1.0; H, 90.4±1.14; HV, 87.9±0.56; HE-B, 112±1.12; HE-A, 102.8±0.37), Bax (B, NC, 100±0; NS, 100.3±0.73; H, 113.7±1.23; HV, 115.9±1.08; HE-B, 105.7±1.48; HE-A, 104.9±1.07), and caspase-3 (C, NC, 100±0; NS, 100.9±0.41; H, 116.9±1.57; HV, 118.5±1.76; HE-B, 107.2±1.63; HE-A (105.9±2.51) antibodies 7 days after hypoxic insult, and the ratio of Bax/Bcl-2 expression (D). Recombinant human erythropoietin (rHuEPO) was administered intraperitoneally at 1,000 IU/kg. NC, Normoxia control group (n=7); NS, Normoxia sham-operated group (n=8); H, Hypoxia group (n=8); HV, Hypoxia+Vehicle group (n=7); HE-B, Hypoxia+rHuEPO-treated group before hypoxic insult (n=8); HE-A, Hypoxia+rHuEPO-treated group after hypoxic insult (n=8). *P<0.05 compared with hypoxia.

Fig. 3
Real-time PCR of Bcl-2 (A; NC, 100±0; NS, 91.4±2.63; H, 80.7±1.1; HV, 77.4±1.39; HE-B, 118.1±1.63; HE-A, 115.7±2.32), Bax (B; NC, 100±0; NS, 100.7±0.51; H, 111.7±0.6; HV, 110.9±1.24; HE-B, 104.2±0.97; HE-A, 98.6±1.35), and caspase-3 (C; NC, 100±0; NS, 116.5±1.23; H, 135.7±2.8; HV, 139.5±1.11; HE-B, 97.3±0.68; HE-A, 102.1±0.74) primers 7 days after hypoxic insult, and the ratio of Bax/Bcl-2 expression (D). Recombinant human erythropoietin (rHuEPO) was administered intraperitoneally at 1,000 IU/kg. NC, Normoxia control group (n=7); NS, Normoxia sham-operated group (n=8); H, Hypoxia group (n=8); HV, Hypoxia+Vehicle group (n=7); HE-B, Hypoxia+rHuEPO-treated group before hypoxic insult (n=8); HE-A, Hypoxia+rHuEPO-treated group after hypoxic insult (n=8). *P<0.05 compared with hypoxia.
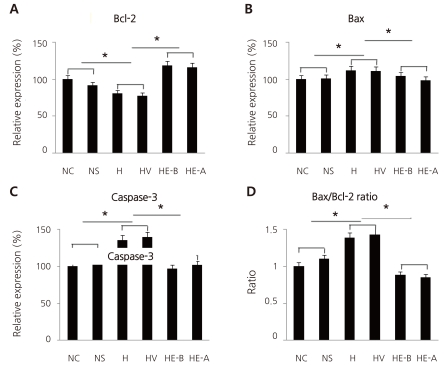
Fig. 4
High-magnification (×200) photomicrograph of brain cortical cell culture from a 19-day-pregnant Sprague-Dawley rat. Recombinant human erythropoietin (rHuEPO) was administered at 1, 10, and 100 IU/mL. A, Normoxia group (N); B, Hypoxia group (H); C, Hypoxia+1 IU/mL rHuEPO-treated group (H+E1); D, Hypoxia+10 IU/mL rHuEPO-treated group (H+E10); E, Hypoxia+100 IU/mL rHuEPO-treated group (H+E100).

Fig. 5
Cell viabilities were measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay. N (100±0), normoxia group; H (34.6±0.7), hypoxia group; H+E1 (84.6±1.72), hypoxia+1 IU/mL recombinant human erythropoietin (rHuEPO)-treated group; H+E10 (61.5±1.14), hypoxia+10 IU/mL rHuEPO-treated group; H+E100 (38.5±0.89), hypoxia+100 IU/mL rHuEPO-treated group. *P<0.05 compared with hypoxia.

Fig. 6
Western blots of Bcl-2 (A; N, 100±0; H, 97.6±0.96; H+E1, 95.2±0.45; H+E10, 102.9±0.75; H+E100, 98.7±0.85), Bax (B; N, 100±0; H, 104.3±0.57; H+E1, 97.7±0.7; H+E10, 91±0.87; H+E100, 84.3±0.54), and caspase-3 (C, N, 100±0; H, 107.8±0.71; H+E1, 99.2±0.63; H+E10, 104.6±0.89; H+E100, 126.9±0.61) in cultured cortical neuronal cells from 19-day-old rat embryos, and the ratio of Bax/Bcl-2 expression (D). Recombinant human erythropoietin (rHuEPO) was administered at 1, 10, and 100 IU/mL. A, Normoxia group (N, n=4); B, Hypoxia group (H, n=4); C, Hhypoxia+1 IU/mL rHuEPO-treated group (H+E1, n=4); D, Hypoxia+10 IU/mL rHuEPO-treated group (H+E10, n=4); E, Hypoxia+100 IU/mL rHuEPO-treated group (H+E100, n=4). *P<0.05 compared with hypoxia.
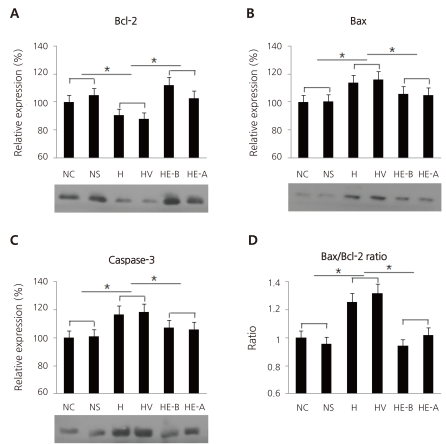
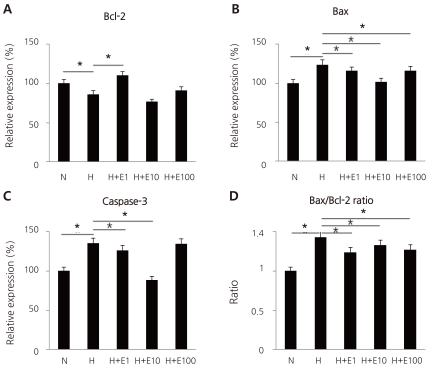
Fig. 7
Real-time PCR of Bcl-2 (A; N, 100±0; H, 86.5±0.84; H+E1, 110.2±2.1; H+E10, 76.8±1.38; H+E100, 91.4±0.98), Bax (B; N, 100±0; H, 123.9±1.31; H+E, 1,115.7±1.6; H+E10, 102.1±1.89; H+E100, 116.5±0.82), and caspase-3 (C; N, 100±0; H, 135.7±0.79; H+E1, 126.6±1.16; H+E10, 88.9±0.69; H+E100, 134.7±0.74) mRNAs in cultured cortical neuronal cells from 19-day-old rat embryos, and the ratio of Bax/Bcl-2 expression (D). Recombinant human erythropoietin (rHuEPO) was administered at 1, 10, and 100 IU/mL. A, Normoxia group (N, n=4); B, Hypoxia group (H, n=4); C, Hypoxia +1 IU/mL rHuEPO-treated group (H+E1, n=4); D, Hypoxia+10 IU/mL rHuEPO-treated group (H+E10, n=4); E, Hypoxia+100 IU/mL rHuEPO-treated group (H+E100, n=4). *P<0.05 compared with hypoxia.